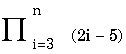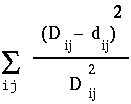Lecture 7, part 2 of 4
| PLNT4610/PLNT7690
Bioinformatics Lecture 7, part 2 of 4 |
|
- the product of function from i to n For comparison, an estimate of the number of protons in the universe is between 1078 and 1082. |
All 15 tree topologies for 5
species
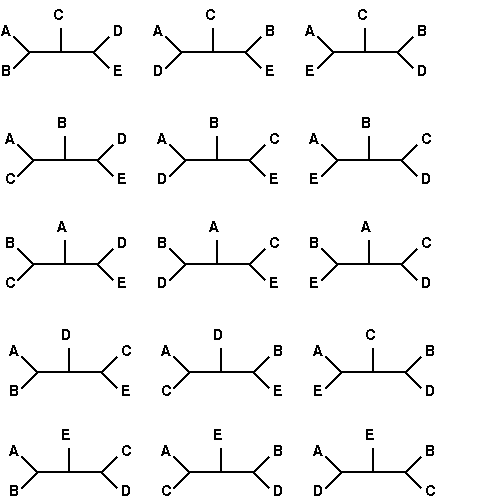
redrawn from
Felsenstein [http://www.cs.washington.edu/education/courses/590bi/98wi/ppt15/sld011.htm
].
Therefore, unless only
a small number of sequences are to be included in a tree,
methods to avoid considering obviously suboptimal trees must be
used to reduce the total number of trees considered.
A phylogenetic
tree is a graph consisting of nodes and branches
Any
phylogenetic tree can be defined by two components:
topology and branch length.
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
DNA scoring methods:
(for in-depth descriptions, see http://home.cc.umanitoba.ca/~psgendb/doc/Phylip/dnadist.html)
transition - purine for purine or pyrimidine for pyrimidine substitution
eg. A -> G, G -> A, C -> T, T -> Ctransversion - purine for pyrimidine, or pyrimidine for purine substitution
eg. A -> C, A -> T, T -> G, T ->A, C -> G, C -> A etc...Transitions occur much more frequently during evolution than transversions.
Protein scoring methods
(for in-depth descriptions, see http://home.cc.umanitoba.ca/~psgendb/doc/Phylip/protdist.html)
Because of the many nuances in working with proteins, there are
many scoring schemes. Most are based on existing PAM or BLOSUM
matrices. One common method is to use Dayhoff's PAM 001 matrix
to score distances. (One PAM unit is defined as the amount of sequence
divergence corresponding to a 1% amino acid replacement rate.) Alternatively, Kimura's
protein distance metric simply uses observed amino acid
frequencies from a protein to approximate a PAM distance:
D = -ln (1 - p - 0.2 p2)
where p is the fraction of amino acids that differ
between two sequences
Using the appropriate
scoring methods, all pairwise distances between sequences are
calculated. For more details on protein scoring matrices, see
the documentation for the Phylip protdist
program.
For example, the PHYLIP documentation gives the example of
a set of 5 short aligned DNA sequences
Alpha AACGTGGCCACATThe corresponding distance matrix using the Kimura 2 parameter model is
Beta ..G..C......C
Gamma C.GT.C......A
Delta G.GA.TT..G.C.
Epsilon G.GA.CT..G.CC
| Alpha | Beta | Gamma | Delta | Epsilon | |
| Alpha | 0.2997 | 0.7820 | 1.1716 | 1.4617 | |
| Beta | 0.3219 | 0.8997 | 0.5653 | ||
| Gamma | 1.4481 | 1.0726 | |||
| Delta | 0.1679 | ||||
| Epsilon |
| B | C | |
| A | 24 | 28 |
| B | 32 |
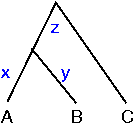
Simultaneous linear equations can be used to calculate the branch lengths:
A to B: x + y = 24Thus with 3 equations and 3 unknowns we can calculate that x = 10, y = 14 and z = 18. These pairwise distances are the shortest distance between each possible pair of nodes.
A to C: x + z = 28
B to C: y + z = 32
Addition of branches is iterative. Branches are added until all
sequences are included in the tree. This is illustrated in the
example below:
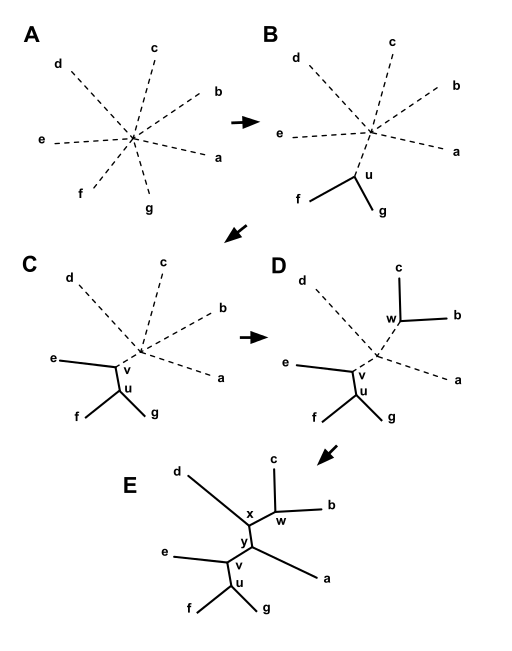
| Starting with a star tree (A), the Q matrix
is calculated and used to choose a pair of nodes for
joining, in this case f and g. These are joined to a newly
created node, u, as shown in (B). The part of the tree shown
as solid lines is now fixed and will not be changed in
subsequent joining steps. The distances from node u to the
nodes a-e are computed from equation (3).
This process is then repeated, using a matrix of just the
distances between the nodes, a,b,c,d,e, and u, and a Q
matrix derived from it. In this case u and e are joined to
the newly created v, as shown in (C). Two more iterations
lead first to (D), and then to (E), at which point the
algorithm is done, as the tree is fully resolved. from https://en.wikipedia.org/wiki/Neighbor_joining |
 |
Advantages
| Fitch and Margoliash showed that different
sets of internal branch lengths could be obtained by
considering alternate trees which moved one or more
branches to different parts of the tree. Consider a
distance matrix for four sequences with pairwise distances
Dij; |
|
||||||||||||||||||||||||||||||
The Neighbor-Joining
tree for these sequences is
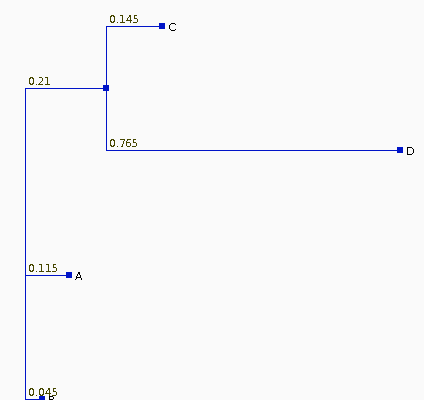
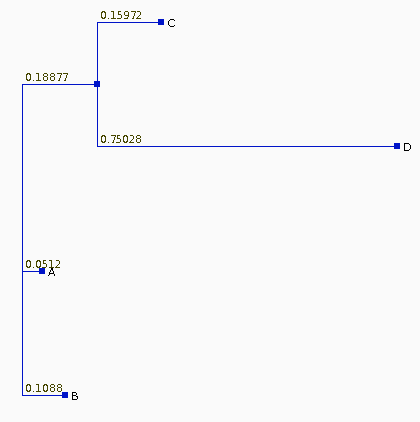
| If we recalculate the pairwise distances dij
from the tree, they are different from the original
distances, as shown at right. The least squares method of Fitch and Margoliash tries different tree topologies, swapping branches among closely-related sequences, and reculating the distances. For each tree considered, a different matrix of distances will be generated (dij). The best tree is defined as that tree which minimizes:
|
|
||||||||||||||||||||||||||||||
| What about
UPGMA? It has been exhaustively demonstrated in the literature, both on theoretical grounds and from phylogenies constructed on strains of known pedigree, that UPGMA is the least robust method. It is based on the assumption that rates of evolution are constant along all branches ie. follows an evolutionary clock. This assumption is almost never valid. Especially with the many choices of far more sophisticated phylogeny inference methods, there is little justification for ever using UPGMA. One point to make is that a comparison of Neighbor-Joining results with UPGMA results could provide a test for the hypothesis that evolution in a given tree is clock-like. As well, UPGMA might be useful for constructing trees where no underlying evolutionary model is assumed. For example, in clustering of genes into groups based on similarities in gene expression patterns, it would be incorrect to assume that genes with similar patterns of expression must in some sense be related in an evolutionary sense. Distance Methods [http://helix.biology.mcmaster.ca/721/outline2/node49.html] Nei M and Roychoudhury AK (1993) Evolutionary Relationships of Human Populations on Global Scale. Mol. Biol. Evol. 10:927-943. Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406-425. |
| PLNT4610/PLNT7690
Bioinformatics Lecture 7, part 2 of 4 |