Lecture 10, part 2 of 2
| PLNT4610/PLNT7690
Bioinformatics Lecture 10, part 2 of 2 |
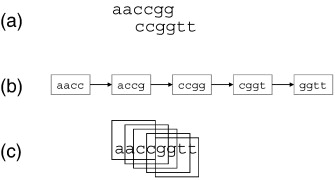
| This
example shows the ideal situation, in which reads
contained no errors. (a) Two overlapping reads (b) A k-mer graph representing all k-mers that overlap the two reads. The graph is a path through connecting the k-mers, to correspond to the overlapping reads. (c) The alignment is a byproduct of graph construction. If you construct a graph through the overlapping k-mers, you implicitly construct an alignment. |
 |
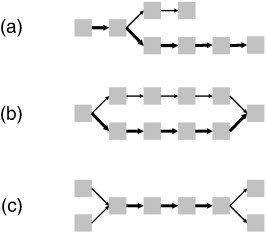
| (a) spur - dead end paths (b) bubble - Two alternate paths, originating at a single k-mer and converging on a single k-mer at both ends. Bubbles are often caused by repeats. Often caused by an error in the middle of a read. (c) frayed rope - two or more divergent paths converge on a unique path, and later diverge. Can also be caused by repeats. |
 |
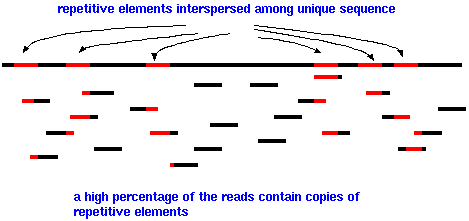
| Eukaryotic
genomes are especially difficult to assemble because so
much of the genome consists of repetitive elements, such
as the AluI family, interspersed among unique DNA. Since
the length of sequencing reads is fairly short, a high
percentage of reads will have part of a repetitive element
at one end. Few reads will completely span a repetitive
element, with unique sequence on either side. |
 |
| While
it is true that repetitive sequence elements do mutate, it
is often difficult or impossible for sequence assembly
software to decide which copy of a repetitive element to
join with any of thousands of other copies that may be
identical or nearly identical to the a given read. Put another way, we don't know where on the chromosome each read came from. That is what we're trying to figure out. The net result is that as a growing contig encounters a repetitive element, there may be no way to extend the contig further. Consequently, most genome assemblies have a relatively small number of large contigs, and a very large number of small contigs, maybe 1000 bp or smaller. |
 |
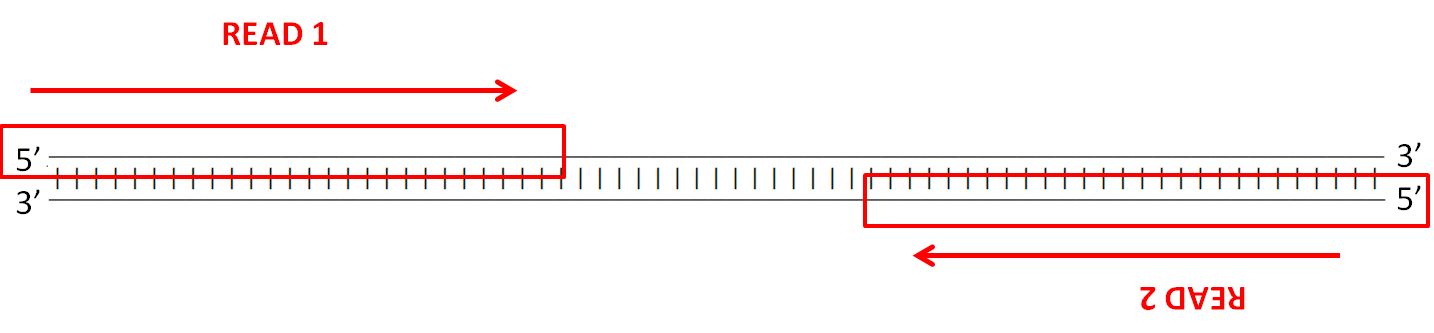
Paired-end reads
| Most sequencing technologies (eg. Illumina,
Ion Torrent etc.) allow the construction of paired-end
libraries, with primer adaptor sites on both ends. For each
fragment two reads are produced. While it is not guaranteed
that the reads will be long enough to overlap each other,
the insert size is known. Insert sizes in current Illumina sequencing typically ranges between 300 and 700 nt. |
 |
Since the size of the
insert is known, algorithms that resolve cases where alternative
paths exist can use the known distance between the paired-end
reads to constrain which path is chosen.
Mate-pair reads
| Mate-pair reads are simply paired-end reads with much longer insert sizes eg. 3000 nt. Again, assembly software can choose paths through the data such that k-mers from one read to its mate-pair must span a distance consistent with the known insert size. | 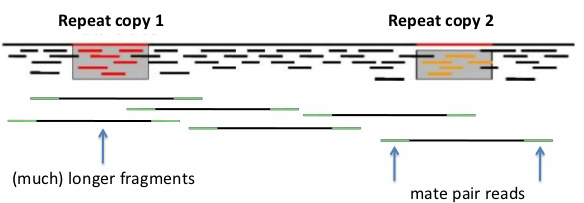 |
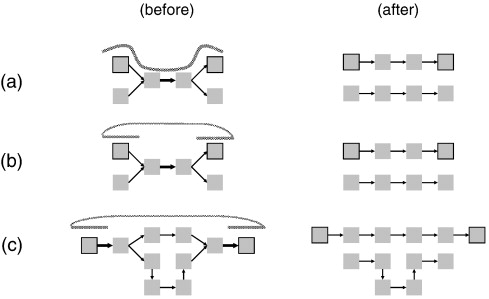
| (a)
When a single read spans a path consisting of several
k-mers, the correct path can be chosen if the read
includes all k-mers in that path. (b) If two reads in a paired-end read overlap terminal k-mers in a frayed-rope ambiguity, the path that includes correct termini can be chosen based on that overlap. (c) Where ambiguous paths exist in a longer path, mate-paired reads can eliminate those paths that do not contain k-mers found in both mate pair reads. Mate pair reads are particularly good at jumping over regions containing repeated sequences longer than the reads. |
 |
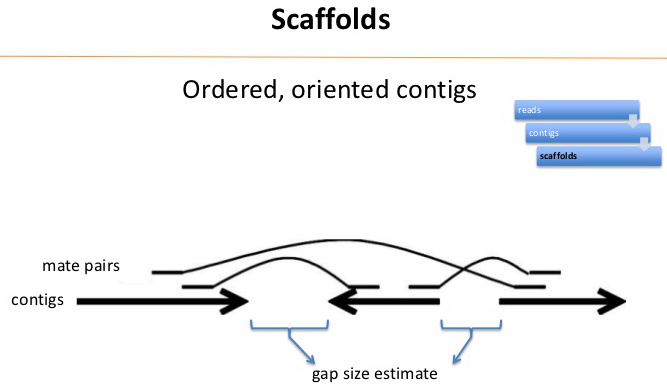
| At
the end of contig assembly process, we are left with a set
of contigs, and a pool of unassigned reads. |
 |
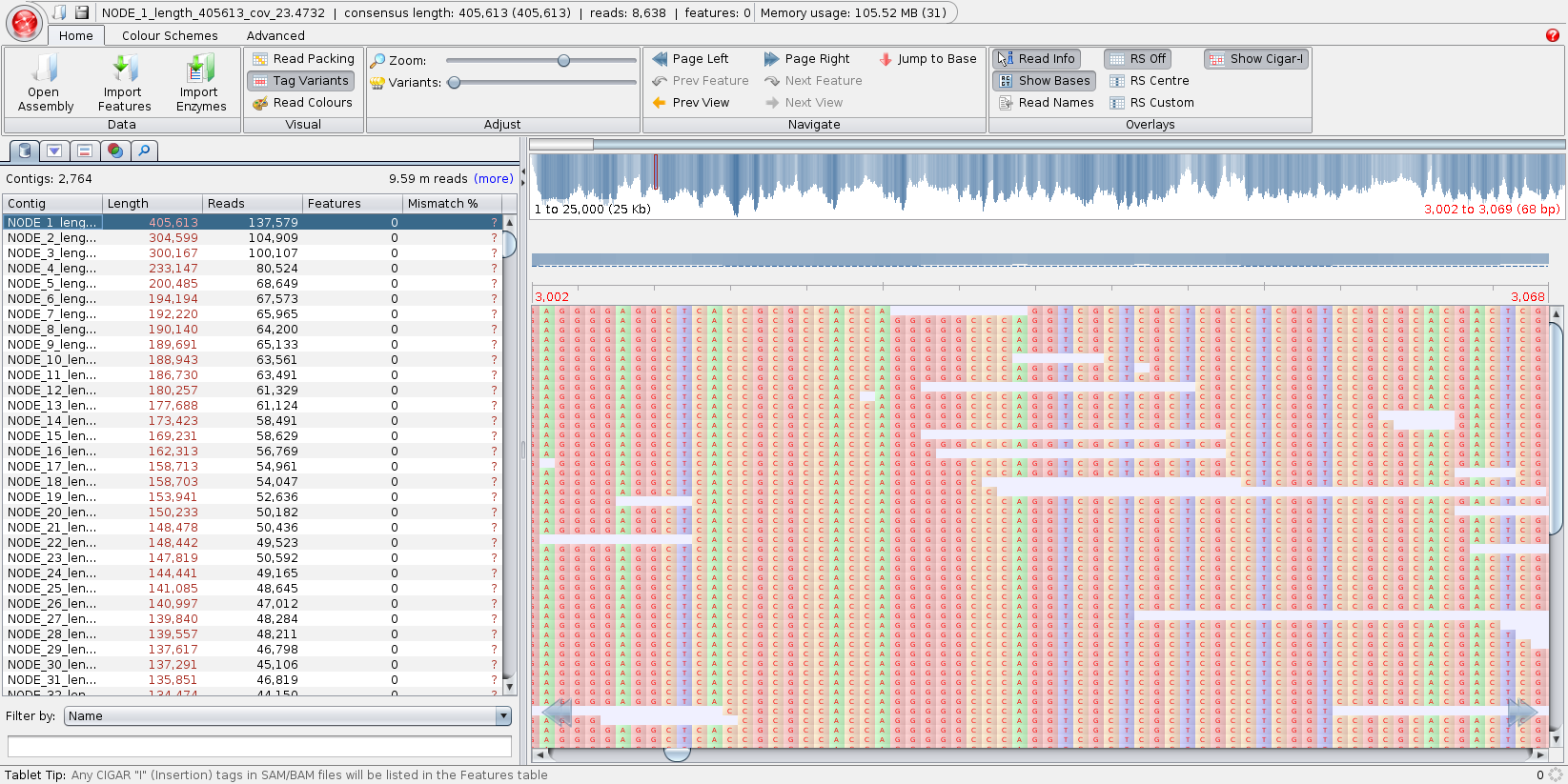
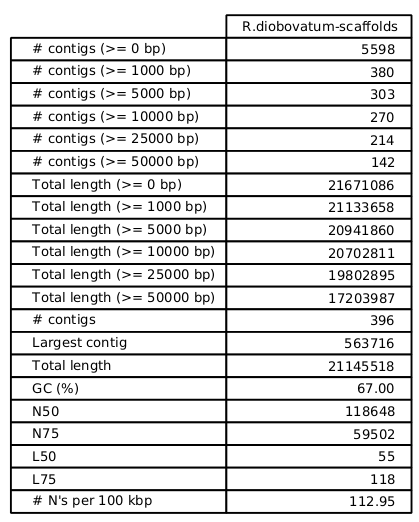
Fakankun I, Fristensky B, Levin DB (2021) Genome Sequence Analysis of the Oleaginous Yeast, Rhodotorula diobovata, and Comparison of the Carotenogenic and Oleaginous Pathway Genes and Gene Products with Other Oleaginous Yeasts. J. Fungi 7:320. https://doi.org/10.3390/jof7040320 Programs such as Quast can generate statistics, summarizing the quality of the assembly. In the ideal, you would like to have each chromosome in a single contig. In practice, with current sequencing technologies, that seldom happens. N50 - Size of the contig such that 50% of the contigs are shorter, and 50% are longer than the N50 contig. The N50 value is the most common single number for evaluating the quality of an assembly. While it is true that the larger the N50, the better, that is not the whole story. It is possible to get misassemblies that create artifactual contigs with high N50 values. |
 |
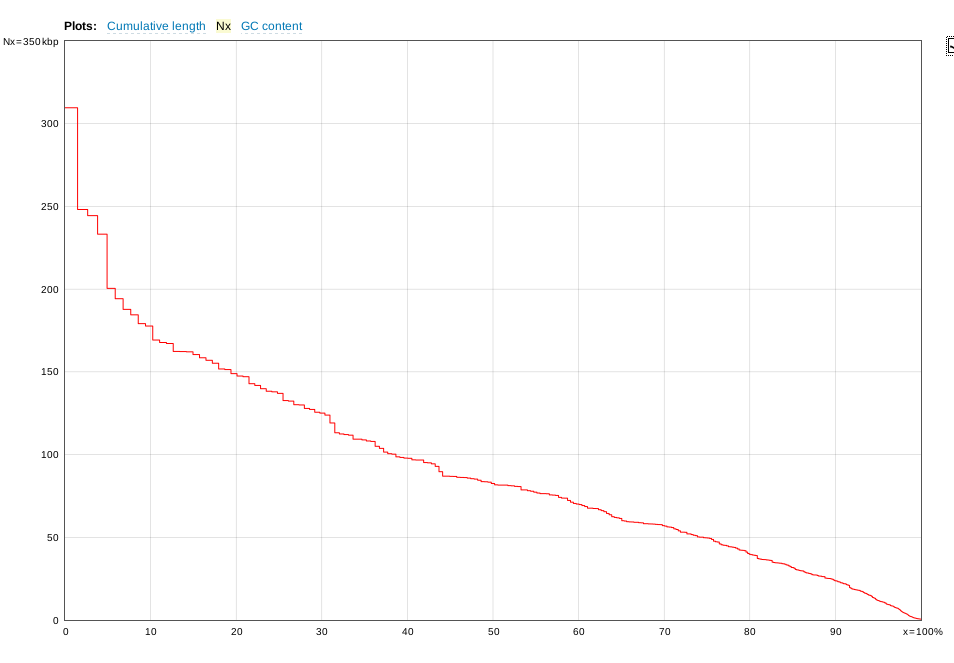
| The
distribution of contigs is generally skewed to a
relatively small number of very large contigs, and a large
number of smaller contigs. An example of an assembly
for a fungal genome of approx. 21,000,000 nt is shown. X axis - cumulative percentage of contigs Y axis - size of contigs |
 |
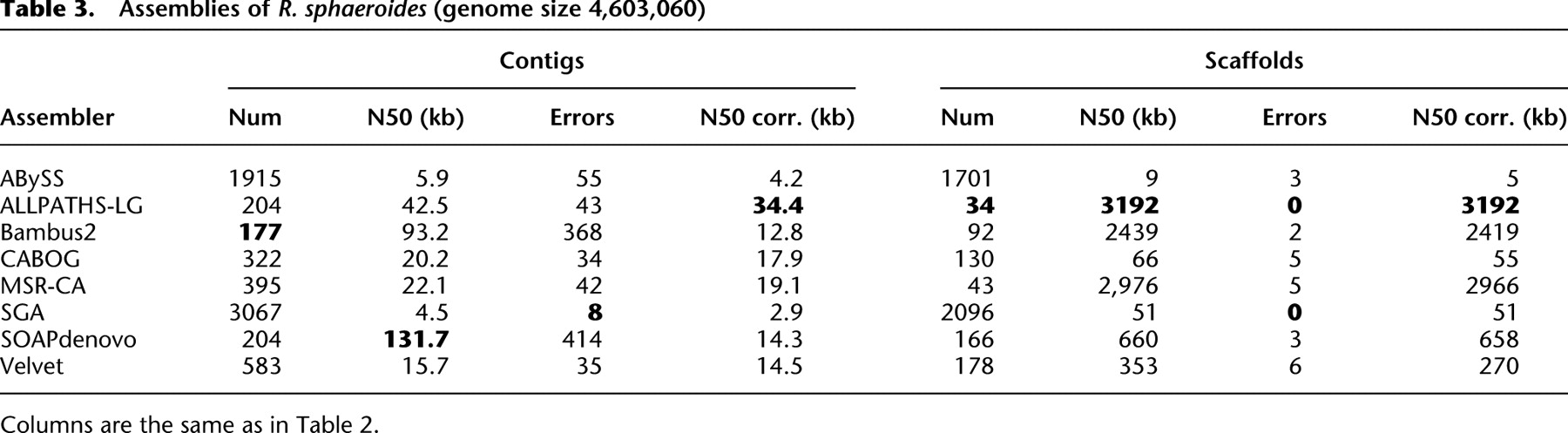
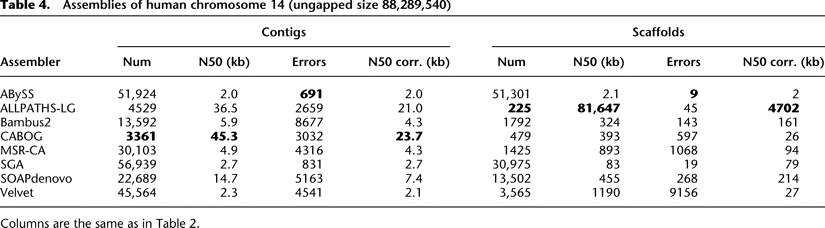
Current state of the art:
|
 Unless otherwise cited or
referenced, all content on this page is licensed under
the Creative Commons License Attribution
Share-Alike 2.5 Canada Unless otherwise cited or
referenced, all content on this page is licensed under
the Creative Commons License Attribution
Share-Alike 2.5 Canada |
| PLNT4610/PLNT7690
Bioinformatics Lecture 10, part 2 of 2 |