TUTORIAL: CREATING DATASETS OF RELATED SEQUENCES |
August 6, 2024 |
TUTORIAL: CREATING DATASETS OF RELATED SEQUENCES |
August 6, 2024 |
Nonetheless, it should
be possible to use 'defensin' as a starting point, and build a
dataset in several steps.
| Note: The sequence dataset constructed in this exercise should be considered a 'first draft'. For example, many of the proteins listed in these GenBank entries are referred to in the annotation as 'defensin-like proteins'. Several rounds of multiple sequence alignment and phylogenetic analysis may be required before a decision can be reached as to which proteins to consider an orthologous group, and which to exclude from the group. |
mkdir dataset
cd dataset
| Launch
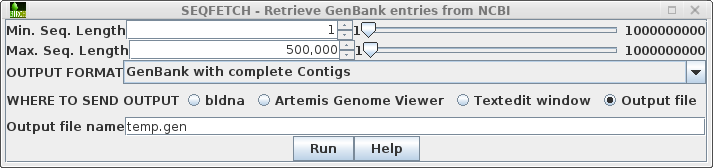
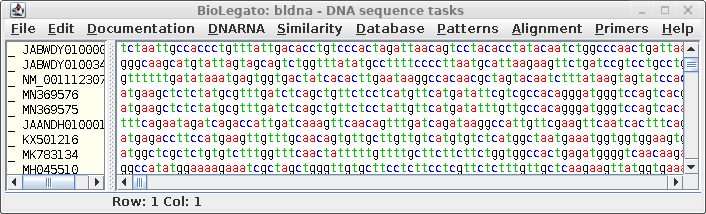
blnci by typing blncbi For working with tabular data, blncbi uses the BioLegato table canvas, that works similar to a spreadsheet. In the Database menu, choose Nucleotide - Query NCBI nucleotide database. |
 |
| A Quick Lesson on Entrez
Query Statements Entrez lets you do highly specific searches by creating search statements in the following form: term [field]
OPERATOR term [field]
where term is a search string, limited to a specific field. The field must be enclosed in square brackets. One or more term-field statements can be joined using the operators AND, OR or NOT, which must be capitalized. See the Entrez Searching Options for more information on writing search statements. |

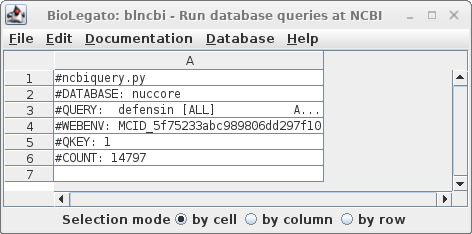
| Click
on Run: Output to a new window, and a new blncbi
window will pop up with the search results. The lines beginning with hash signs are pseudo-comments that summarize the results. The COUNT line shows that 14797 entries were found in which the word defensin appears somewhere in a GenBank entry. |
 |

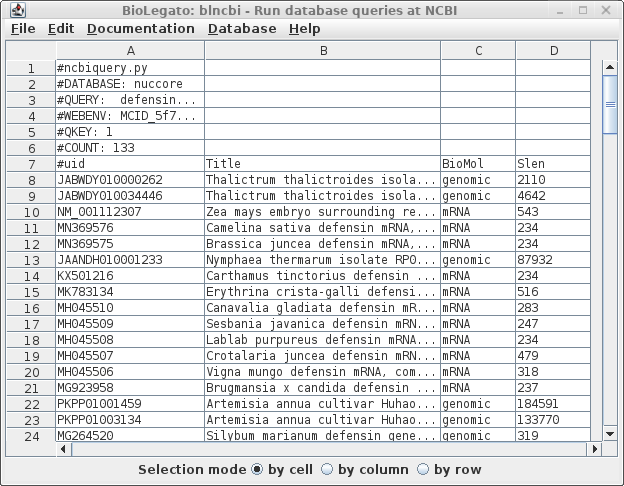
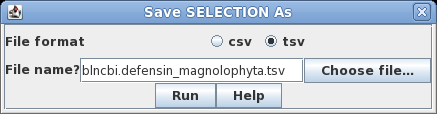
This search report can be saved to a file for future user by choosing Edit --> Select All, and then choosing Save SELECTION As. Type a descriptive filename, such as blncbi.defensin_magnoliophyta.tsv, and set the file format to tsv so that blncbi can read this file if we want to use the results later. |
 |
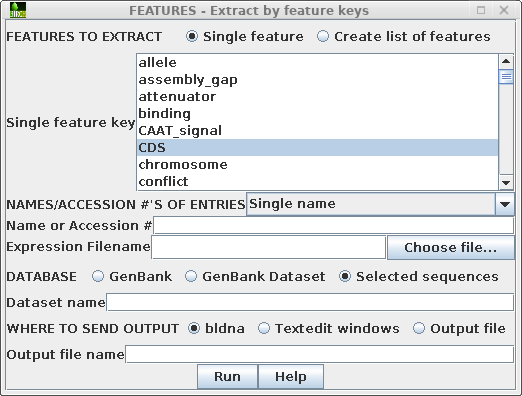
Next, choose Database --> FEATURES - extract
by feature keys.
|
By default, the 'Single feature key' menu is set to 'CDS'. Stay on the "from selected" tab to tell FEATURES to extract subsequences from the sequences you have selected. (If you wanted t FEATURES would first retrieve sequences from GenBank, use the "from NCBI" tab. The "from local files" tab could be used to extract features from a local GenBank dataset.)The menu should look like this: |
 |
| The
coding sequences will appear in a new bldna window |
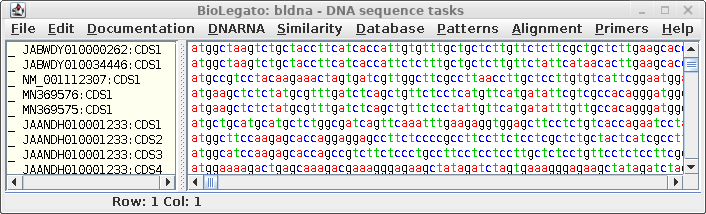 |
Consistent with the
fact that protein coding regions have been extracted from the
total DNA sequences, all of the CDS regions begin with 'atg'.
These can be translated in one step by selecting all sequences
and choosing DNARNA --> Ribosome. By default,
ribosome sends output to a new blprotein window.
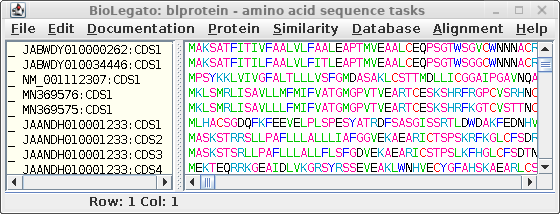
To find other proteins
related to defensins, we can run BLAST searches of defensin
proteins versus translated GenBank sequences. Our first choice
of a test sequence will be AY313169, a defensin homologues from
the barrel medic (Medicago truncatula).
(Hint: You can find for
any sequence in a bioLegato instance window using Edit --> Select by name.)
Once you have selected AY313169, open Database --> NCBI TBLASTN.
Since we expect to have greater than 100 sequences by the end of the process, it is not realistic to work at the 5% or 1% statistical level. To ensure that no hits are found due to random chance, we will consider sequences with E > 10-11 not significant. Set #matches expected to 1 x 10-11 to ensure that only signficant matches are found. The choice is somewhat arbitrary, but in practice, hits would later be pruned by other criteria. |
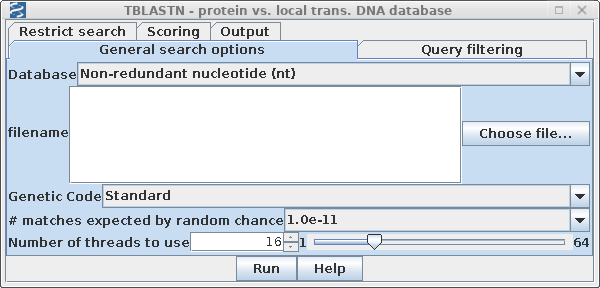 |
| Finally, because the nt is one of the largest NCBI databases, the search may take awhile. Open the Output tab and choose Notify of completion by email. | 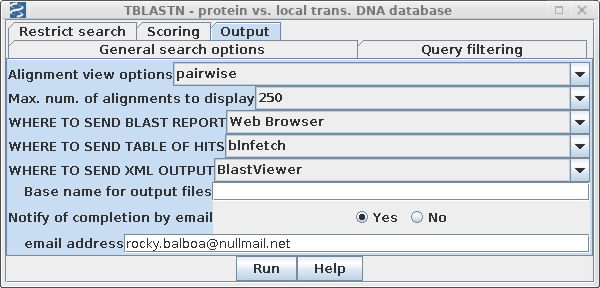 |
Output will
appear in three windows.
| The
HTML BLAST report gives a human-readable list of hits,
followed by the best alignment for each hit. An excerpt is
shown an right. The list contains links both to the
GenBank entry for the hit at NCBI, as well as a link to
the alignment. |
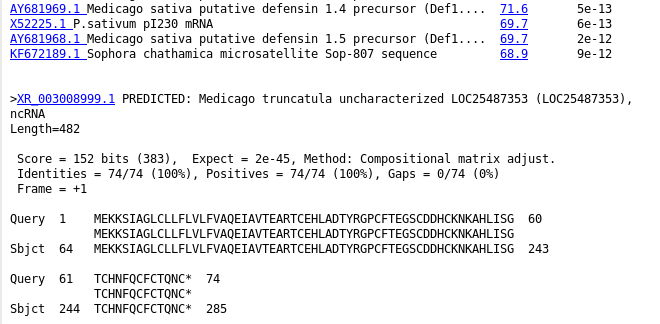 |
| A
tab-separated value (tsv) file with more detailed hit
information is displayed in blnfetch, a BioLegato
interface specialized for displaying and retrieving hits
from nucleotide databases. |
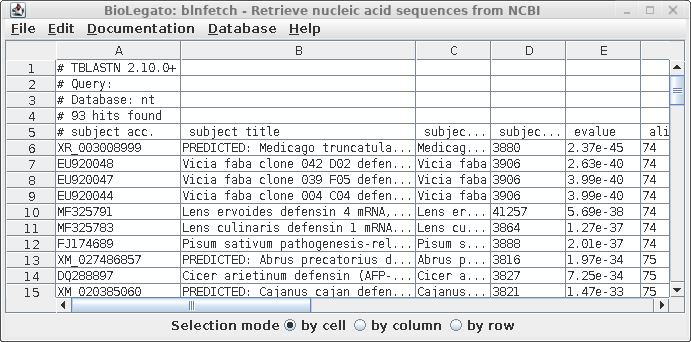 |
blnfetch is a simple
spreadsheet that makes it easier to sift through large
numbers of hits, and includes sort and extract functions
which are valuable for selecting subsets of hits eg.
hits from a particular species. Save the BLAST
report using the file name AY313169.tblastn.htm (File
--> Save Page As).
Before saving the
tsv file, we need to make sure that there aren't any
sequences too large for our purposes eg. in a classroom
tutorial. This example illustrates the value of having
BLAST results in a table. Note that the length of each hit
found is given in the field labeled 'subject length'
(column I).
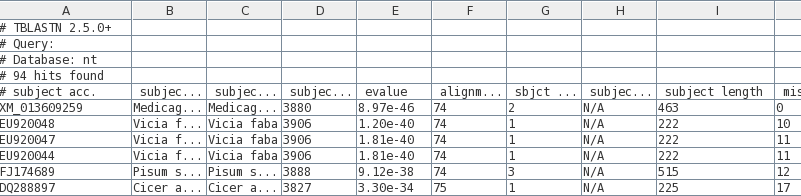
| We
can sort the table based on the 'subject length' by
Edit --> Select All, and then Edit --> BLSORT. Since column I is the ninth column, set the first sort key to column 9, and sort order to descending. |
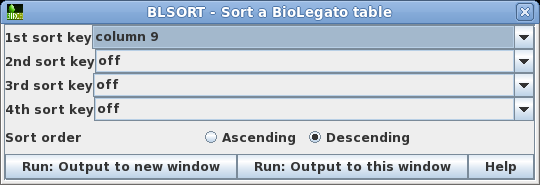 |
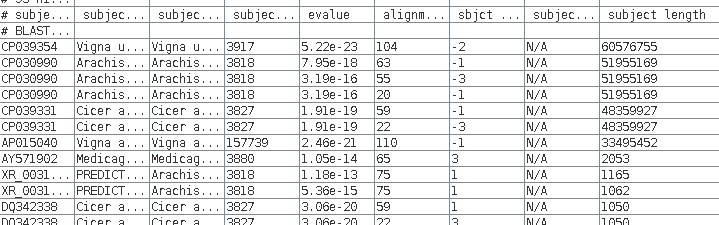
We see that 7
sequences over 33 million bases long, so for the purposes
of saving disk space we want to delete those from the
file.
Make sure the Selection mode at bottom is set to "by
row", select these rows, and then delete using Edit
--> Delete Rows.
The data from the new blnfetch window can be saved by selecting all hits (Edit --> Select All), and then File --> Save SELECTION As AY313169.tblastn.tsv.
Output: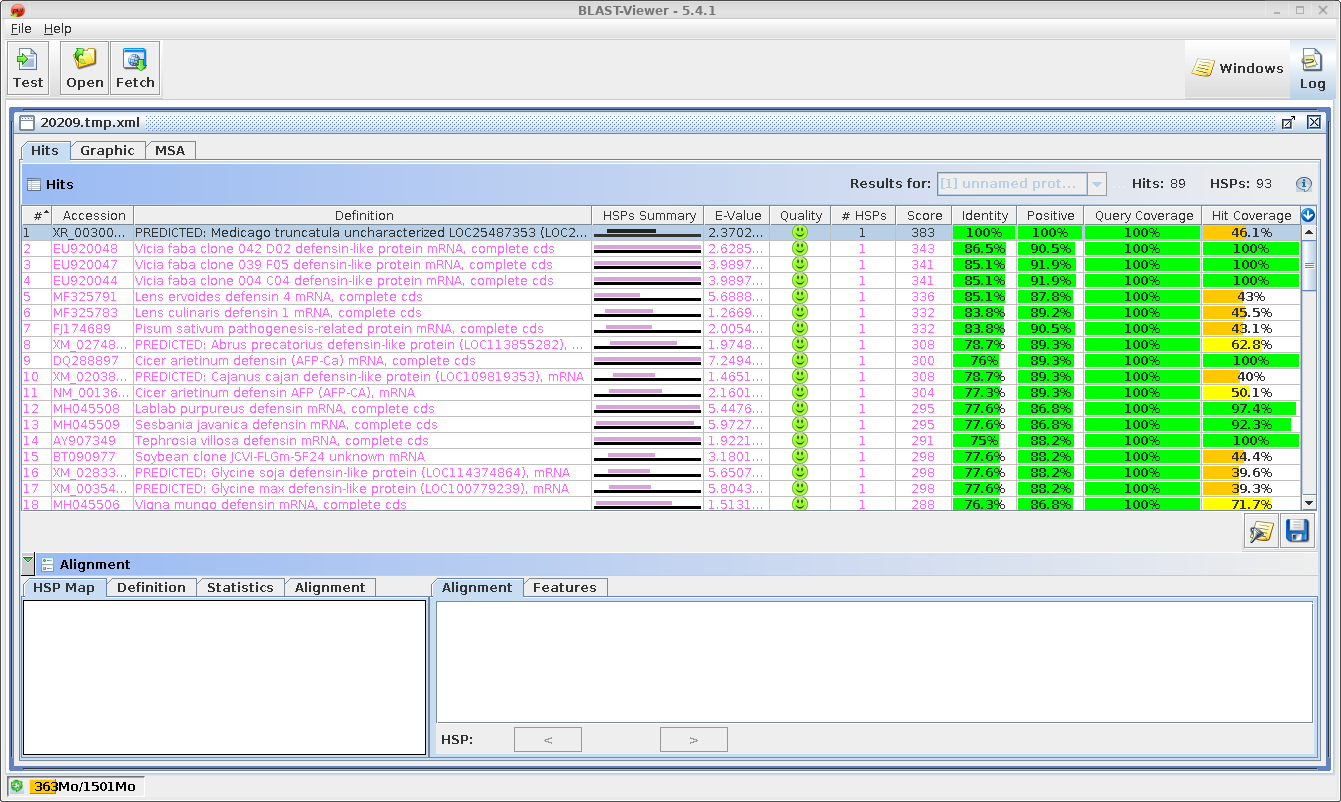
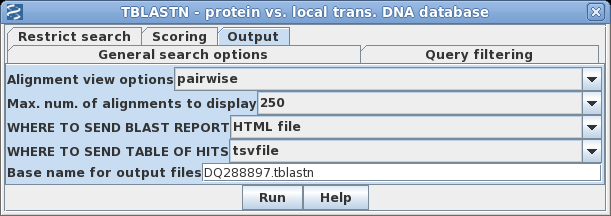
| To
save a few steps, you can send the output directly to
files, bypassing the popup windows. Go to the Output tab, and set the BLAST REPORT to HTML file, and the TABLE OF HITS to tsvfile. Fill in a base name to be used for the two output files as shown at right. |
 |
grep -v '#' -h *.tblastn.tsv | cut -f1 > tblastn.accThe -h option is necessary because there is more than one input file. By default, when grep has more than one input file, it will print the name of each file for each line printed. The -h option suppresses printing of filenames. Note that because this is a tsv file (ie. tab-separated value) the delimiter used by the cut command must be the TAB character! Fortunately, cut defaults to use TAB as a delimiter, so we don't need to specify a delimiter in this particular case.)
cat temp.acc tblastn.acc | sort | uniq > all.acccat effectively combines these files into a single output stream. Sort, as the name implies, sorts the lines in the ouput stream, and uniq eliminates duplicate lines from a sorted file. See the manual pages for each command to get a better idea of what they do (eg. man sort, man uniq).
Note that the combined lengths of temp.acc and tblastn.acc add up to 552, and all.acc is only 434. This is because the tblastn searches will often find the same sequences, so there is some redundancy in the .acc files.
{mars:/home/psgendb/tutorials/bioLegato/dataset}wc -l *.acc
500 AB089942.tblastn.acc
401 all.acc
102 AY313169.tblastn.acc
85 DQ288897.tblastn.acc
409 tblastn.acc
123 temp.acc
1620 total
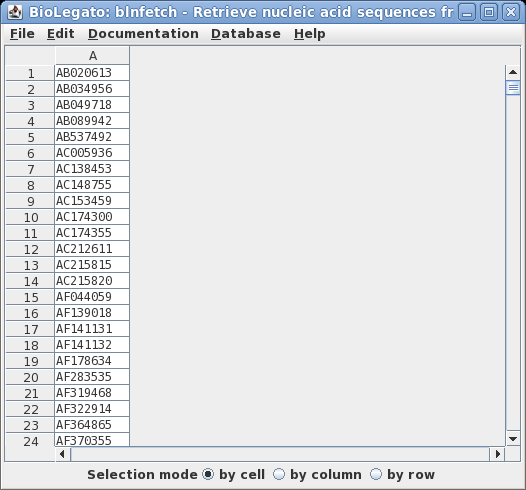
| Next, we want
to retrieve the corresponding sequences. The
BioLegato interface blnfetch is specialized for
retrieving entries from GenBank, using ACCESSION
numbers. Type blnfetch all.acc |
 |
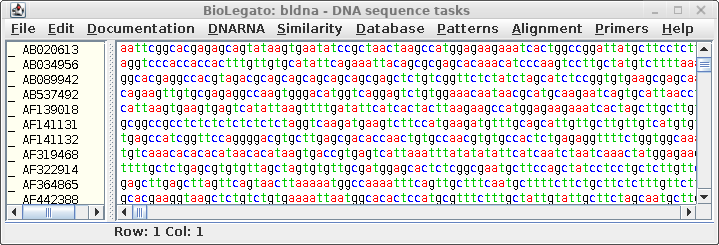
| Save
the
sequences in this file by choosing File --> Save All As, and call the
file all.CDS.fsa. Make sure you choose the Fasta format
for saving the file. This file will be read later by
bldna. Note: Do NOT use Export Foreign Format. This function uses readseq to interconvert formats. However, readseq will truncate sequence names if they are longer than 16 characters. The added ':CDSxx' puts these names over the 16 character limit. Consequently, when there are multiple coding sequences in a given entry, several sequences will end up with the same name. |
 |
As more and more large genomic fragments are entered into GenBank, FASTA searches will find sequences that match only a small part of the total fragment. That is, the gene you are using as a query sequence will be only one of many genes on the fragment. Thus, FEATURES will generate all protein coding sequences (CDS) for all genes on each fragment.
| We can create a small
database of all proteins translated from
the CDS sequences, and then search with
FASTA to identify which encode
defensins. As was done previously, translate all CDS sequences to proteins by choosing Edit --> Select All and using DNARNA --> Ribosome. |
 |
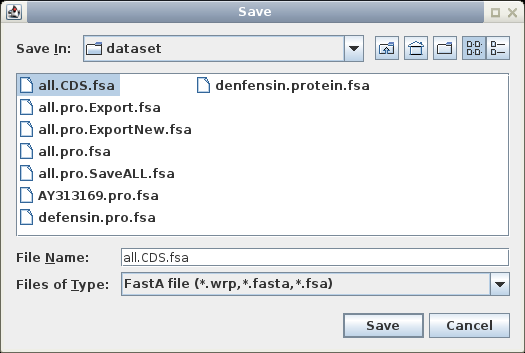
In the new window,
select all sequences (Edit
-- Select All), and choose File --> Save ALL As, and call the
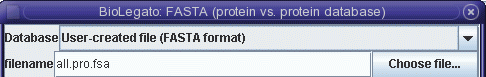
file all.pro.fsa.
The file will contain the protein sequences from all of the CDS sequences.
We can use the same three defensin
sequences as used in the original database search to
search all.pro.fsa to find defensins. To start, find the
AY313169 amino acid sequence using Edit --> Select sequence
by name
Next, choose Database --> Fasta
(protein vs. protein database).
| This
time,
instead of searching GenBank, search the data set in
all.pro.fsa. |
|
|
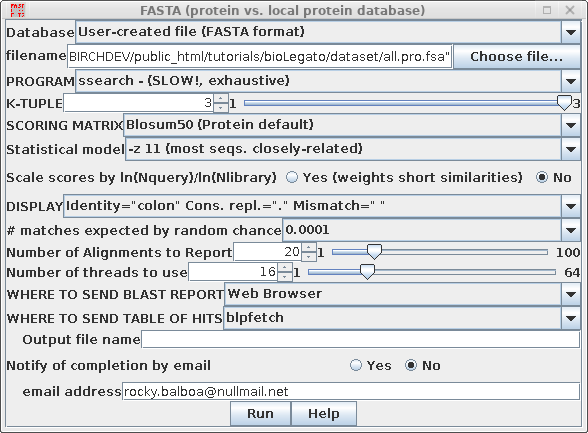
| Use
ssearch, which constructs a rigorous Smith/Waterman
alignment for each pairwise comparison. |
 |
|
| Set the E value, which is the cut off for
listing hits. I this case, hits will only be included in
the output if they have a probability of matching by
random chance of less than 0.0001. |
||
| By default, the FASTA
programs calculate statistics assuming that significant
hits will occur with only a very small fraction of
sequences in the database. This assumption is invalid with
the all.pro.fsa dataset. Setting the -z 11 option
tell FASTA to calculate E values for a population in which
most sequences are closely-related. If you do not set this option, you will miss most of the homologous coding sequences! |
 |
|
This is what it should look like with all the settings:
Repeat the process for the other two sequences. There
should now be three blpfetch windows with the results
from each of the searches:
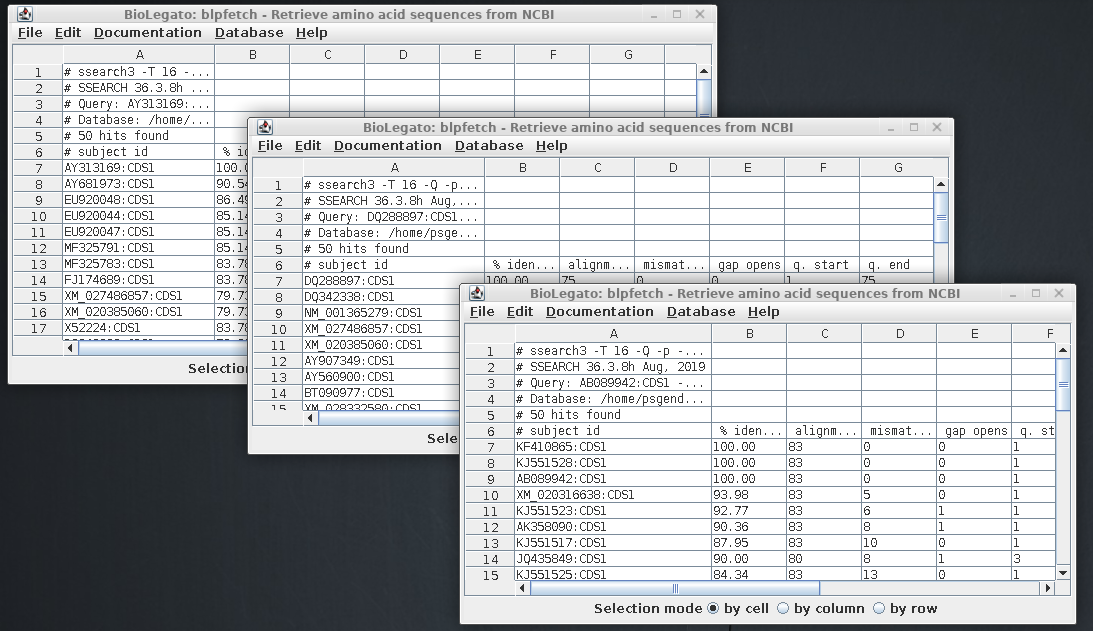
The ssearch results in the three
output files tell us which CDS sequences are defensins.
The next step is to combine the results into a single
window, so that we can create a non-redundant list of
sequences.
Simply choose any of the three blpfetch windows to
combine all hits, and copy the hits from the other two
windows into it. As an example, we'll use the blpfetch
window for which the query was AY313169 for combining
hits.
Go to the blpfetch window in which the Query on line 3 says "DQ288897" and choose Edit --> Select All, and then Copy out. This copies all lines to the blpfetch clipboard. Next, go to the blpfetch window for Query: AY313169, and choose Edit --> Paste in. Repeat this process to copy all hits from the AB089942 window to the AY313169 window.
| To
verify that that multiple sets hits are now in the
AY313169 window, scroll down. You should see the end of
one list, and the beginning of another, as indicated by
the comment lines beginning with hash marks (#). Optional: Save the list as a file. Edit --> Select All. File --> Format: tsv Name: defensin.pro.hits.tsv |
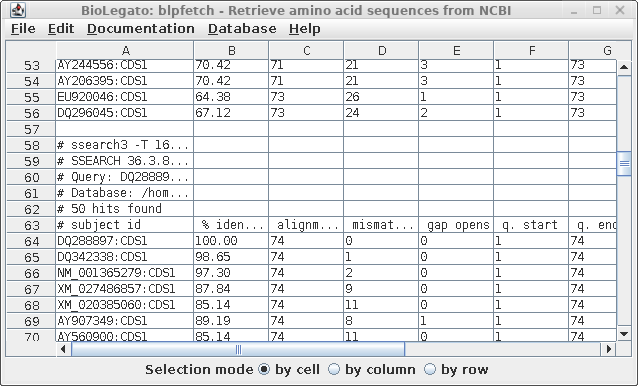 |
| Next,
we need to get the list of hits to paste into the bldna.
Go back to the blpfetch window for AY313169, containing
the combined list of hits, and set the Selection mode at
the bottom to "by column". Next click in column A. The
window should look similar to the one at right. Copy the selection into the clipboard: Linux: ctrl-C Mac: command-C |
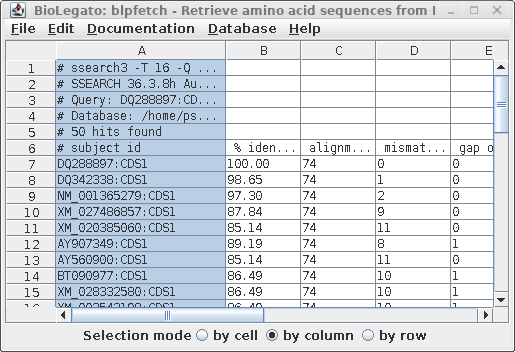 |
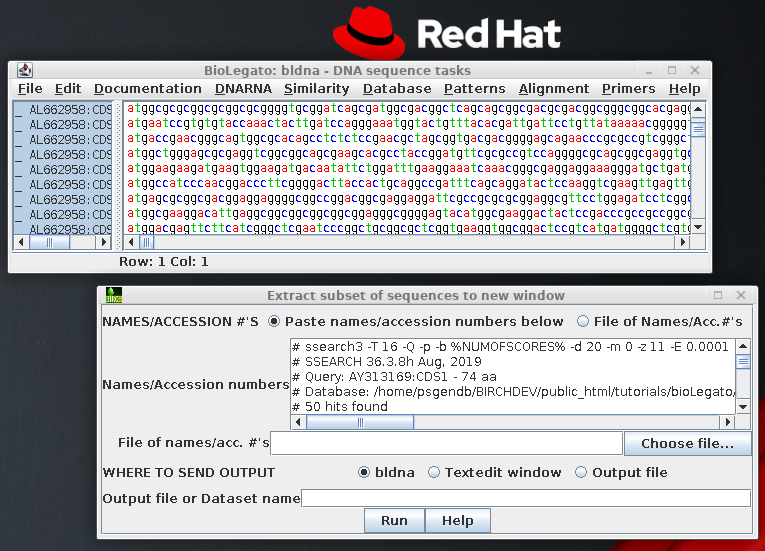
Save these sequences in
File --> Save As, under the name defensin.CDS.fsa. Make sure to write the file in
Fasta format.
This illustrates an important point
regarding database searches. Most database searches will
only find the BEST alignment between a query sequence
and a sequence in the database. In
the case where several homologous genes are present in
a single database entry, only the best match with the
query sequence is likely to be reported, and the
others ignored! If we hadn't been careful, we
would have missed 3 out of the 4 defensin-related genes
in AC005936.
To be complete, let's also save the
amino acid sequences corresponding to these CDS
sequences. Choose DNARNA --> Ribosome:
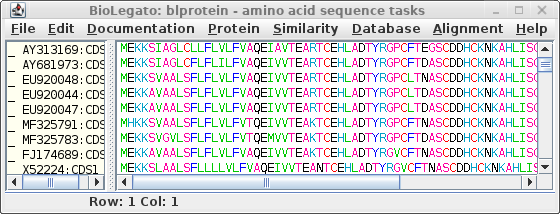
Save these sequences as defensin.pro.fsa.
|
The dataset is complete! It is now ready
for creating multiple sequence alignments, and from
there, for phylogenetic analysis. |
The comparison of the forward strand of AC005936 with the opposite strand of CDS9 shows no compelling similarities, which may not be surprising considering the fact that CDS9-12 were all on the forward strand, according to the Features Table.
The clustering of members of the defensin gene family on one strand, within several kilobases is probably evidence that these genes have been duplicated by unequal crossing over in recent evolutionary time.