
 VMOdes for Windows VMOdes for Windows
VMOdes for Windows VMOdes for Windows Virtual Molecular Orbital description Program
(Current version available is A 7.2)
The most popular modern quantum-chemical packages like Gaussian, HyperChem, Spartan, GAMESS, and ADF generate numerical description of molecular orbitals in output files. However, the main question for chemists is the contribution of different shells, atoms, and fragments to the molecular orbital of interest. So, how do you do this? You may spend a lot of time calculating such contributions using a calculator or develop your own program to give the information you are looking for. VMOdes is an alternative way. It is easy to use. It is freeware. It will be upgraded in the future (Gaussian 09 capability is coming soon). You can download it from this web-page or follow links provided at www.gaussian.com!
Part 1. Program capabilities and limitations.
Part 2. Examples of using VMOdes.
Part 3. List of files distributed with VMOdes program and installation notes.
Program capabilities and limitations.
VMOdes (Virtual Molecular Orbital description Program) is a small program for calculating contribution of the selected atomic orbital(s) into molecular orbital(s). Selected atomic orbitals can represent atomic shell (for instance metal d-manifold, dz2 orbital, px orbital, etc.), atoms, or group of atoms. VMOdes produces publication quality tables with molecular orbital analyses.
We developed this program in order to analyze the chemical interactions between atoms or fragments in molecular systems. We were interested in developing this program because in general, most of the quantum-chemical software packages are not capable of atomic orbital contribution analysis of molecular orbitals.
The current version of VMOdes processes output files from the following quantum-chemical programs:
Output files from restricted, restricted-open, and unrestricted calculations at semi-empirical, ab initio , and DFT levels are processed by VMOdes.
In the current version of VMOdes only the most popular for the molecular orbital description c^2 population analysis is available (P. Ros and G. C. A. Schuit, Theoret. Chim.Acta (Berl.) 1966, 4, 1-12).
Operating System: Windows Me, 2000, XP, Windows 7 (I stopped supporting Windows 95, 98, & NT OS)
System Requirements: Pentium processor with 32MB RAM is minimum, but the more the better. For output files about 200Mb in size you will need ~2Gb of RAM.
Gaussian 03 and 03W output files are processed with VMOdes.
Windows 7 (Home & Professional) is included in the list of VMOdes supported OS.
GAMESS ( USA ) output files are processed with VMOdes;
For unrestricted wavefunctions it is now possible to analyse Alpha and Beta orbital sets separately.
Maximum number of orbitals: 1500
Maximum number of groups in a subunit: 15
Maximum number of subunits: 7
Description:
VMOdes does not work well with UNIX/LINUX Gaussian 98 and GAMESS (USA) output files.
Solution:
Delete all the UNIX/LINUX generated path lines before line Entering Link 1 = (for Gaussian) or **** (for GAMESS (USA)) output files.
Description:
Some times VMOdes does not respond when it runs under Windows 95.
Solution:
Close VMOdes using the Task Manager. VMOdes output file is already generated.
Gaussian 09 support.
ADF and Spartan output files will process with VMOdes.
The number of subunits will be increased to 10.
Citation of the VMOdes is the part of the license agreement.
Cite this program as: VMOdes Program, Revision A 7.2
V. N. Nemykin, P. Basu; University of Minnesota Duluth and Duquesne University; 2001, 2003, 2005.
Examples of using VMOdes.
Before you start, make sure that:
- Your output file contains all the necessary information on the molecular orbitals;
- Your data.txt file contains all the necessary information on the shell(s), atom(s), or group(s) of interest.
To make sure that your output file contains all the necessary information on the molecular orbitals use the following settings:
Software Settings
Gaussian Pop=Full
GAMESS no specific settings
HyperChem QuantumPrintLevel = 1 (or higher, I typically use QuantumPrintLevel = 9)
All information on subunits of interest should be located in data.txt file. This is an ASCII file which can be edited by using any ASCII file editors (Notepad, WordPad, FAR manager, etc.). The format of the data.txt file for VMOdes should be the following:
Line 1: Number of subunits (any integer number between 1 and 7)
Line 2: Subunit specification: i) number of shell(s), atom(s), or group(s) in subunit, ii) name of subunit following coma, iii) first and the last basis function values for each shell(s), atom(s), or group(s) in subunit.
Line 3: Specification of next subunit (if necessary).
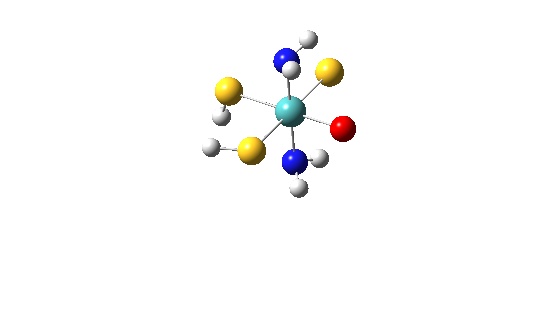 The data.txt file listed below is for a MoOS(SH)2(NH3)2 calculation using Gaussian 98:
The data.txt file listed below is for a MoOS(SH)2(NH3)2 calculation using Gaussian 98:
7
1 Molybdenum, 1 37
1 Molybdenum d-orbitals, 16 37
1 Oxygen atom, 38 44
1 Sulfur atom (Mo=S), 45 51
2 SH ligands, 52 62 74 76
2 NH3 ligand, 63 73 77 80
1 Total, 1 80
In total 7 subunits are considered.
The first one is for all AOs of Molybdenum (basis functions from 1 to 37).
The second subunit is the d-metal manifold of Molybdenum (basis functions from 16 to 37).
The third subunit is the Oxygen atom (basis functions from 38 to 44).
The fourth subunit is the Sulfur atom (basis functions from 45 to 51).
The fifth subunit are the two SH groups consisting of two subgroups. The first one, is two Sulfur atoms (basis functions from 52 to 62) while the second one is the Hydrogen atoms (basis functions from 74 to 76).
The sixth subunit is the NH3 ligands consists of one Nitrogen atom (basis functions from 63 to 73) and three Hydrogens (basis functions from 77 to 80).
The seventh subunit is total. If you have less that 7 subunits considered, it is recommended to use the total subunit in order to be sure that the sum of all basis functions will give you 100% of the MOs contribution (VMOdes is a freeware and still may contain bugs!). For more detailed discussion on data.txt file, see full examples below.
As soon as you have unzipped VMOdes.zip file, start VMOdes*.exe. As soon, as you start it, the message box (I hope so :-) ) will appear. Click OK. Now you should get the main window of the VMOdes program:
 .
.
Click on the "File Open" button or use the "File/Open" menu. Select and open files as usual in Windows interface:
 .
.
Choose Run/Ci Square Method from menu:
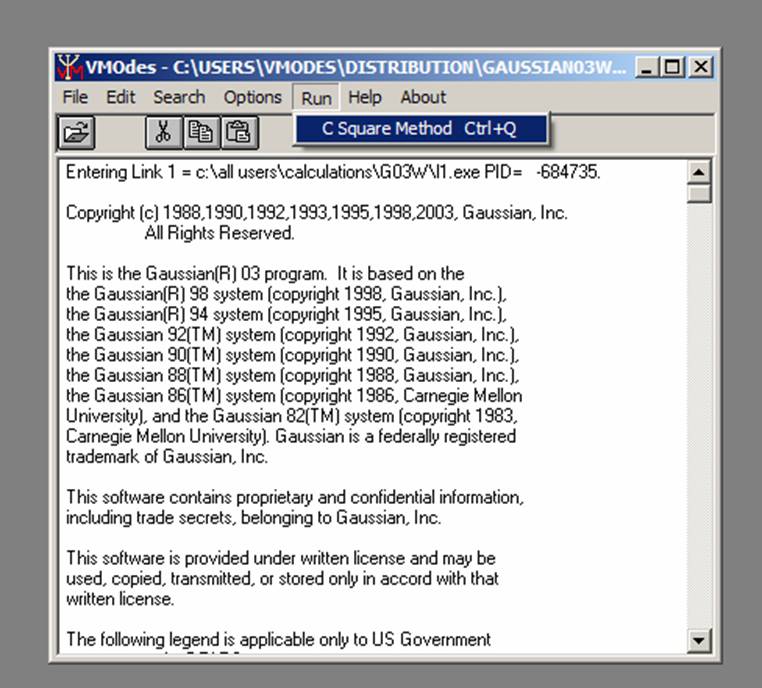 .
.
The new window will appear:
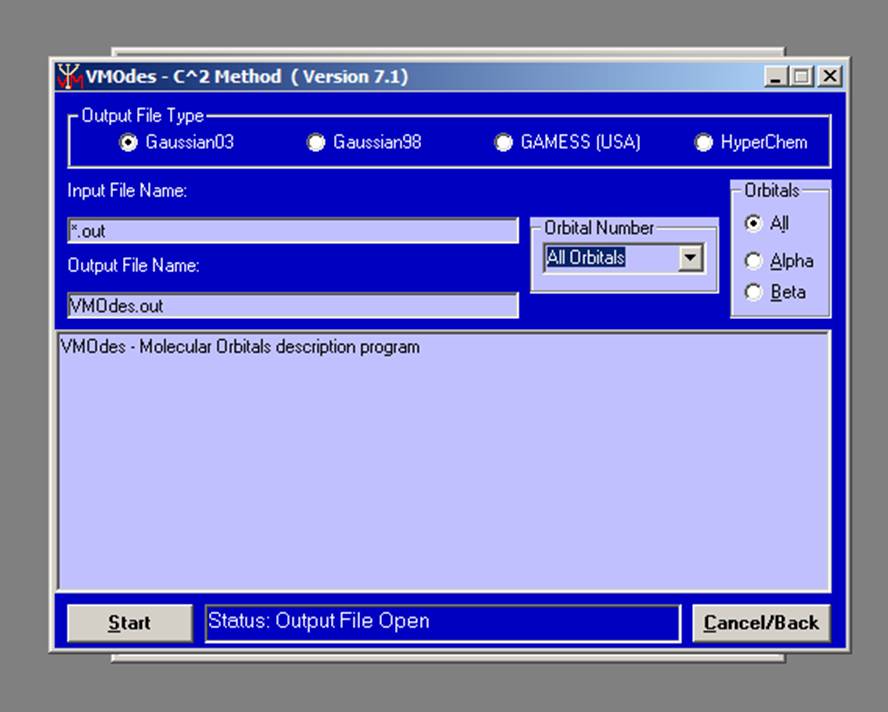 .
.
Next, choose the output file type ( Gaussian 98, Gaussian 03, GAMESS ( USA ), or HyperChem ) from the upper menu. Choose the orbital(s) of interest. You may choose between All orbitals (all orbitals will be considered), 5x5 (only 5 highest occupied and 5 lowest unoccupied orbitals will be considered), 10x10 (only 10 highest occupied and 10 lowest unoccupied orbitals will be considered), 20x20 (only 20 highest occupied and 20 lowest unoccupied orbitals will be considered), or individual orbital (from 1 up to 1500):
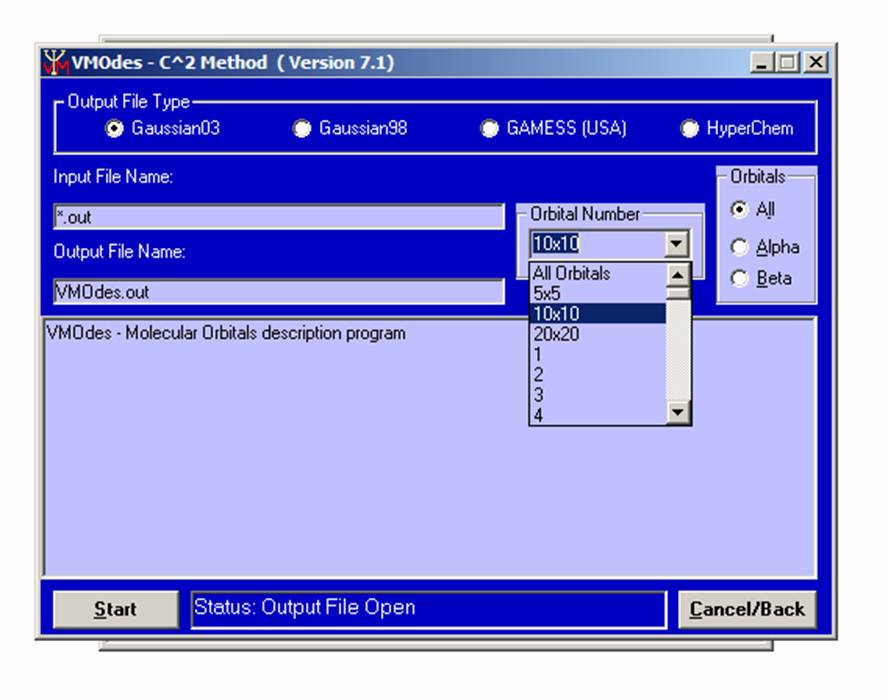 .
.
Next, choose orbital set (alpha or beta). This option is only active for unrestricted calculations. You are all set.
Press the Start button (or Alt+S) and type the VMOdes output file name in the box which will appear:
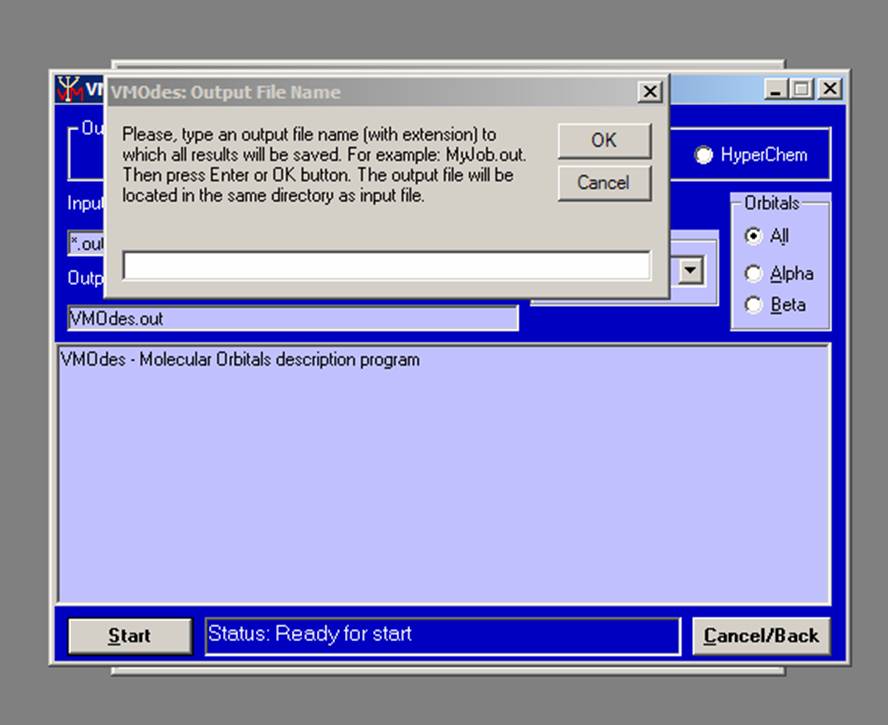 .
.
When all calculations are done, you will see the Status: Calculation Complete message and output information (only for small files!) in the output window:
 .
.
Now you may terminate the VMOdes program or click on the Cancel/Back button and load the next output file. Below you will find a few examples, which you may test with VMOdes program (please rename data-*-example.txt files to data.txt).
FULL EXAMPLE 1 shows the Gaussian output file for a methane molecule calculated at the B3LYP level using 6-31G(d) basis set.
The part of the Gaussian output file looks like:
1 2 3 4 5
(A1)--O (A1)--O (T2)--O (T2)--O (T2)--O
EIGENVALUES -- -11.20980 -0.94056 -0.54367 -0.54367 -0.54367
1 1 C 1S 0.99565 -0.19375 0.00000 0.00000 0.00000
2 2S 0.02830 0.35807 0.00000 0.00000 0.00000
3 2PX 0.00000 0.00000 0.00000 0.00000 0.43425
4 2PY 0.00000 0.00000 0.43425 0.00000 0.00000
5 2PZ 0.00000 0.00000 0.00000 0.43425 0.00000
6 3S -0.01314 0.36696 0.00000 0.00000 0.00000
7 3PX 0.00000 0.00000 0.00000 0.00000 0.23009
8 3PY 0.00000 0.00000 0.23009 0.00000 0.00000
9 3PZ 0.00000 0.00000 0.00000 0.23009 0.00000
10 4XX -0.00204 0.01013 0.00000 0.00000 0.00000
11 4YY -0.00204 0.01013 0.00000 0.00000 0.00000
12 4ZZ -0.00204 0.01013 0.00000 0.00000 0.00000
13 4XY 0.00000 0.00000 0.00000 0.03600 0.00000
14 4XZ 0.00000 0.00000 0.03600 0.00000 0.00000
15 4YZ 0.00000 0.00000 0.00000 0.00000 0.03600
16 2 H 1S 0.00004 0.13373 0.16821 0.16821 0.16821
17 2S 0.00243 0.03807 0.14014 0.14014 0.14014
18 3 H 1S 0.00004 0.13373 -0.16821 0.16821 -0.16821
19 2S 0.00243 0.03807 -0.14014 0.14014 -0.14014
20 4 H 1S 0.00004 0.13373 0.16821 -0.16821 -0.16821
21 2S 0.00243 0.03807 0.14014 -0.14014 -0.14014
22 5 H 1S 0.00004 0.13373 -0.16821 -0.16821 0.16821
23 2S 0.00243 0.03807 -0.14014 -0.14014 0.14014
Let's say we would like to calculate carbon, carbon s-orbitals, and hydrogen atom contributions to each MO. In this case, the data.txt file may look like the following:
4
1 Carbon, 1 15
2 Carbon s orbitals, 1 2 6 6
1 Hydrogen atoms, 16 23
1 Total, 1 23
A VMOdes output file will look like:
Input Gaussian file:
C:\All Users\Calculations\Qbasic\MyPrograms\SDI-1\e2_02.out
Output VMOdes file: test.out
Full point group is: TD
Number of Basis Functions = 23
5 Alpha electrons 5 Beta electrons
Group 1 is: Carbon
This group consists of 1 subunit. The Basis Functions range is:
1 15
Group 2 is: Carbon s orbitals
This group consists of 2 subunits. The Basis Functions range is:
1 2
6 6
Group 3 is: Hydrogen atoms
This group consists of 1 subunit. The Basis Functions range is:
16 23
Group 4 is: Total
This group consists of 1 subunit. The Basis Functions range is:
1 23
All MO's will be printed.
Molecular Orbitals Indexes, Energies, and Group Contributions
Orbital Group Number
_________________________ ____________________________________
Number Index Energy,eV 1 2 3 4
1 (A1)--O -305.037 100.0 100.0 0.0 100.0
2 (A1)--O -25.594 79.5 79.5 20.5 100.0
3 (T2)--O -14.794 55.9 0.0 44.1 100.0
4 (T2)--O -14.794 55.9 0.0 44.1 100.0
5 (T2)--O -14.794 55.9 0.0 44.1 100.0
6 (A1)--V 6.941 64.2 64.2 35.8 100.0
7 (T2)--V 8.846 30.4 0.0 69.6 100.0
8 (T2)--V 8.846 30.4 0.0 69.6 100.0
9 (T2)--V 8.846 30.4 0.0 69.6 100.0
10 (T2)--V 19.912 85.7 0.0 14.3 100.0
11 (T2)--V 19.912 85.7 0.0 14.3 100.0
12 (T2)--V 19.912 85.7 0.0 14.3 100.0
13 (T2)--V 31.988 34.8 0.0 65.2 100.0
14 (T2)--V 31.988 34.8 0.0 65.2 100.0
15 (T2)--V 31.988 34.8 0.0 65.2 100.0
16 (A1)--V 33.628 60.0 59.8 40.0 100.0
17 (A1)--V 35.683 88.3 88.2 11.7 100.0
18 (E)--V 52.860 100.0 0.0 0.0 100.0
19 (E)--V 52.860 100.0 0.0 0.0 100.0
20 (T2)--V 70.137 60.7 0.0 39.3 100.0
21 (T2)--V 70.137 60.7 0.0 39.3 100.0
22 (T2)--V 70.137 60.7 0.0 39.3 100.0
23 (A1)--V 125.281 96.2 56.0 3.8 100.0
FULL EXAMPLE 2 shows the Pc GAMESS (USA) output file for a methylene (1A1 singlet state) molecule calculated at RHF level using STO-3G basis set.
The part of the Pc GAMESS (USA) output file looks like:
1 2 3 4 5
-11.0013 -0.8395 -0.5271 -0.3016 0.2280
A1 A1 B1 A1 B2
1 C 1 S 0.993035 -0.216249 0.000000 0.133125 0.000000
2 C 1 S 0.030638 0.657554 0.000000 -0.610829 0.000000
3 C 1 X 0.000000 0.000000 0.528825 0.000000 0.000000
4 C 1 Y 0.000000 0.000000 0.000000 0.000000 1.000000
5 C 1 Z 0.006480 0.169457 0.000000 0.782815 0.000000
6 H 2 S -0.007445 0.273533 -0.458157 0.213567 0.000000
7 H 3 S -0.007445 0.273533 0.458157 0.213567 0.000000
Let's say we would like to calculate the carbon and hydrogen atom contributions to each MO. In this case, the data.txt file may look like the following:
3
1 Carbon, 1 5
1 Hydrogen atoms, 6 7
1 Total, 1 7
A VMOdes output file will looks like:
Input GAMESS (USA) file:
C:\All Users\Calculations\Qbasic\MyPrograms\SDI-1\Symm-rhf.out
Output VMOdes file: test.out
Full point group is: CNV
The order of the principal axis is: 2
Number of Basis Functions = 7
Number of Electrons = 8
Number of Alpha Electrons = 4
Number of Beta Electrons = 4
Group 1 is: Carbon
This group consists of 1 subunit. The Basis Functions range is:
1 5
Group 2 is: Hydrogen atoms
This group consists of 1 subunit. The Basis Functions range is:
6 7
Group 3 is: Total
This group consists of 1 subunit. The Basis Functions range is:
1 7
All MO's will be printed.
Molecular Orbitals Indexes, Energies, and Group Contributions
Orbital Group Number
_________________________ ___________________________
Number Index Energy,eV 1 2 3
1 A1 -299.363 100.0 0.0 100.0
2 A1 -22.844 77.2 22.8 100.0
3 B1 -14.343 40.0 60.0 100.0
4 A1 -8.207 91.7 8.3 100.0
-----Unoccupied orbitals-----
5 B2 6.204 100.0 0.0 100.0
6 A1 18.741 53.5 46.5 100.0
7 B1 19.805 46.8 53.2 100.0
FULL EXAMPLE 3 shows the HyperChem 6.02 output file for the ammonia molecule calculated at PM3 level.
The part of the HyperChem 6.02 output file looks like:
Mol. Orbital 1 2 3 4 5 6
Symmetry: 1 A1 1 E 1 E 2 A1 3 A1 2 E
Eigenvalue -27.61762 -14.76440 -14.76440 -9.67861 2.79812 4.95296
S N 1 0.76519 0.00000 0.00000 0.37169 -0.52567 -0.00000
Px N 1 0.04791 0.65326 -0.07465 -0.30355 -0.14490 -0.08318
Py N 1 0.02818 0.02742 0.68444 -0.17854 -0.08523 0.69972
Pz N 1 0.12959 -0.24746 -0.12123 -0.82110 -0.39195 -0.12140
S H 2 0.36268 0.49547 -0.30880 -0.14562 0.42496 0.30685
S H 3 0.36268 -0.51516 -0.27469 -0.14562 0.42496 0.26340
S H 4 0.36268 0.01969 0.58349 -0.14562 0.42496 -0.57026
Let's say we would like to calculate nitrogen, nitrogen p-orbitals, and hydrogen atom contributions to each MO. In this case, the data.txt file may look like the following:
4
1 Nitrogen, 1 4
1 Nitrogen p orbitals, 2 4
1 Hydrogen atoms, 5 7
1 Total, 1 7
A VMOdes output file looks like:
Input HyperChem file:
C:\All Users\Calculations\Qbasic\MyPrograms\SDI-1\chem-NH3-rhf.log
Output VMOdes file: test.out
Number of electrons = 8
Number of alpha electrons: 4
Number of beta electrons: 4
Number of basis functions = 7
Group 1 is: Nitrogen
This group consists of 1 subunit. The Basis Functions range is:
1 4
Group 2 is: Nitrogen p orbitals
This group consists of 1 subunit. The Basis Functions range is:
2 4
Group 3 is: Hydrogen atoms
This group consists of 1 subunit. The Basis Functions range is:
5 7
Group 4 is: Total
This group consists of 1 subunit. The Basis Functions range is:
1 7
All MO's will be printed.
Molecular Orbitals Indexes, Energies, and Group Contributions
Orbital Group Number
_________________________ ____________________________________
Number Index Energy,eV 1 2 3 4
1 1 A1 -27.618 60.5 2.0 39.5 100.0
2 1 E -14.764 48.9 48.9 51.1 100.0
3 1 E -14.764 48.9 48.9 51.1 100.0
4 2 A1 -9.679 93.6 79.8 6.4 100.0
-----Unoccupied orbitals-----
5 3 A1 2.798 45.8 18.2 54.2 100.0
6 2 E 4.953 51.1 51.1 48.9 100.0
7 2 E 4.953 51.1 51.1 48.9 100.0
Full point group is: C3V
List of files distributed with the VMOdes program
VMOdes*.exe current version of VMOdes program
License.dat license file (should be updated by request)
Help.txt help file
data-Gaussian-example.txt example of data.txt file for Gaussian job
data-GAMESS-example.txt - example of data.txt file for GAMESS (USA) job
data-HyperChem-example.txt - example of data.txt file for HyperChem job
Gaussian-example.out example of Gaussian output file
GAMESS-example.out example of GAMESS (USA) output file
HyperChem-example.log example of HyperChem output file
Installation of the VMOdes for Windows:
Just simply send an e-mail to: Victor.Nemykin@umanitoba.ca and include the following information:
a) Your name and (if you are student) name of your supevisor
b) Name of the University or company you are working for
c) Contact address & e-mail.
I will use this information only for the VMOdes users/updates database.
Once the information received, you will get the secret code, which is necessary for unzipping VMOdes. Good luck!
© 2004-2017 Victor Nemykin, All Rights Reserved
Last Updated October 02, 2017