BioLegato:
|
June 29, 2022 |
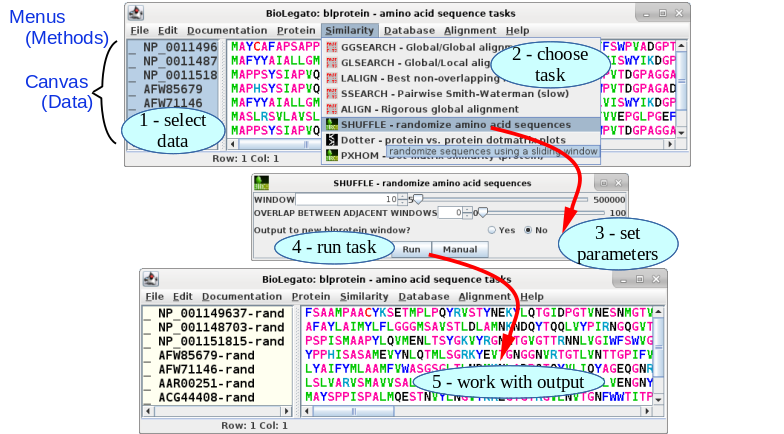
All tasks in bioLegato are done in five steps:
BioLegato:
|
June 29, 2022 |
|
|
    |
|