TUTORIAL: FINDING AND RETRIEVING SEQUENCES FROM NCBI |
Jan. 29, 2020 |
TUTORIAL: FINDING AND RETRIEVING SEQUENCES FROM NCBI |
Jan. 29, 2020 |
| cd tutorials mkdir findseq cd findseq |
go into tutorials directory create a directory called findseq go into the findseq directory |
 . The
BIRCH launcher will appear
. The
BIRCH launcher will appear
| A
chooser will appear on your screen asking for the name of
the directory in which you wish to work. Choose 'tutorials/findseq'
and click on Open. |
 |
| blncbi
will appear on the screen. blncbi has several functions
for searching NCBI, and results are displayed in a
spreadsheet panel. |
 |
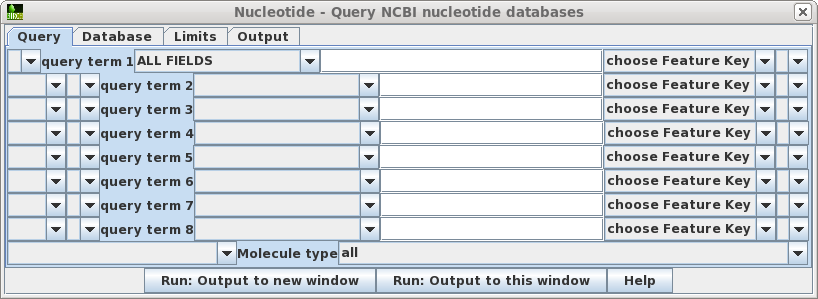
| Query
- Allows you to build a query of up to 8 search terms. For
each term, choose the field to limit the search, and type
in the value. If you choose the Feature Key field, also
choose the Feature Key to search. For example, choosing
the 'intron' feature key would limit retrievals to only
those sequences for which introns are annotated. Pull-down widgets let you group terms with parentheses, and join terms using AND, NOT or OR. At the bottom of the window, you cal also limit searches to a particular molecule type eg. tRNA, rRNA, ncRNA etc. |
 |
| What you need
to know about databases All information in a database is organized into fields. Each field holds a value. For example, if you had a database of people, it might look something like this:
There are three fields in each record: FirstName, LastName, and Phone. Each record has a unique value for each field. (Think of a field name as a variable from algebra.) Any database search program will allow you to search for records which have specific values for one or more fields. All entries in which the field(s) match the specified value are returned. For searching the NCBI databases, the Query tab lets you specify values for one or more fields, and then retrieves entries which match those values. |
| Database
- Sets the database to search. |
 |
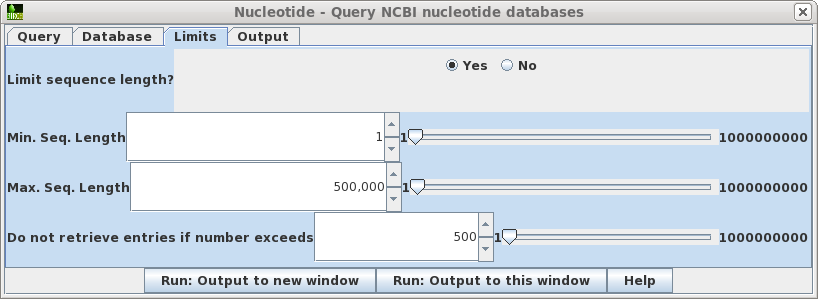
| Limits
- Allows you to retrieve only hits in a particular size
range, and to increase or decrease the number of hits
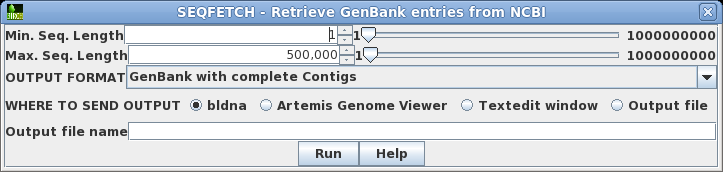
retrieved. Where you know the approximate size of the sequence you want, setting a narrow Min.-Max range can limit a search that would otherwise have hundreds of thousands of hits to a small enough number that you can view them in the output. In such cases, you might increase the maximum hits retrieved to, say, 5000. If you sort the hits by size or even title, you can often quickly scan by eye to find the sequences you really want. |
 |
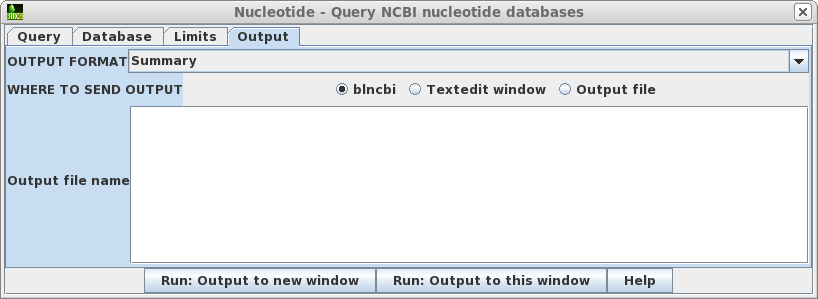
| Output
- Allows you to specify how the output is saved or
formatted. By default, the format is Summary, which
returns output as a table. Although you could change the
format to GenBank, the Summary is usually best, since you
could always retrieve GenBank entries from the Summary
itself. By default, Summary output goes to a new blncbi window, which lets you screen the output, and retrieve selected sequences. If you wish the output to directly to a fiel, choose Output file, and make sure to type in an output filename eg. results.tsv. The .tsv file extension indicates that the Summary output is in TAB-separated value format, which can be read directly by blncbi, or any spreadsheet program. |
 |
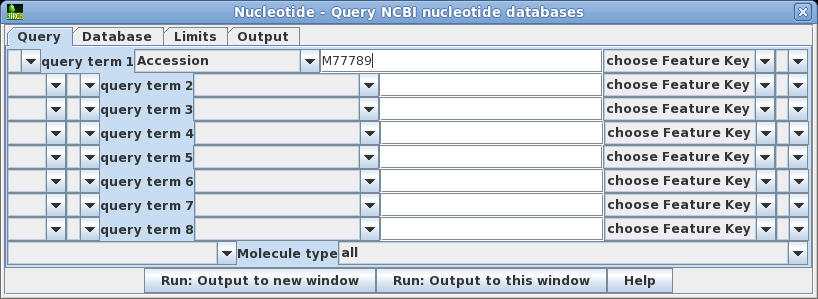
| Choose
Database --> Nucleotide which will open the
query builder. The query builder lets you create query
statements which connect keywords with relations such as
AND, OR, NOT and parentheses. You can also choose specific
databases to search, set parameters limiting things such
as sequence length or number of hits to retrieve, and
where to send the output. Set query term 1 to search the Accession field of GenBank entries for the Accession number M77789. Click on Run: Output to new window. |
 |
|
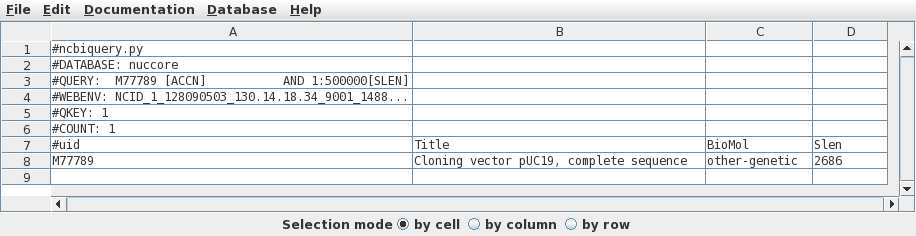
Since there is only one hit, we want to retrieve this one.
Click on M77789 in column A, and then choose Database
--> Seqfetch. By default, seqfetch will retrieve
results to bldna, a BioLegato application for working with
DNA sequences. This is usually the best choice, since
bldna can always save your sequences from bldna, or open
them for viewing in programs such as a text editor or the
Artemis Genome Viewer. Click on Run to retrieve your
sequence. |
 |
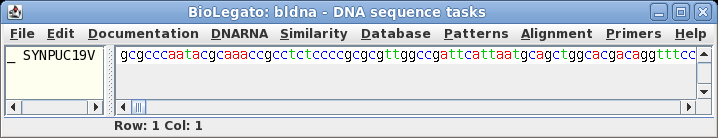
| The
sequence is retrieved from NCBI to a bldna window. |
 |
In these tutorials, we'll see that all
tasks run through BioLegato fall into four basic steps:
|
| Select
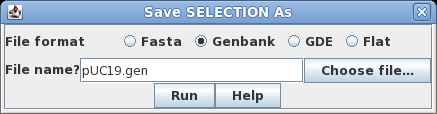
the sequence by clicking on its name, SYNPUC19V. Choose File
--> Save SELECTION AS. To give the file a name
that is more descriptive than the Accession number, let's
call it pUC19.gen. To preserve all sequence annotation,
set the file format to GenBank. Click on Run
to save. |
 |
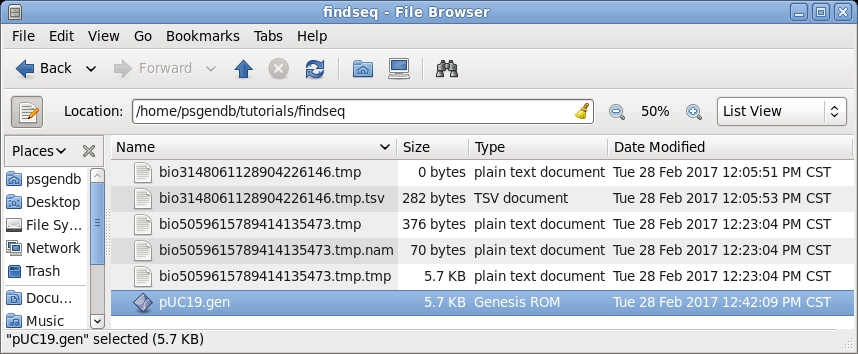
| In
the file manager (finder on Mac) you should now see this
sequence in your findseq directory. (Files whose names begin with 'bioxxxx' are temporary files created by BioLegato. These should automatically be deleted when BioLegato terminates.) |
 |
| To
view your sequence in a text editor, you could either
click on pUC19.gen in the file manager, or from bldna, File
--> View Sequences. The pull-down menu lets you
choose which sequence format you wish to view. For
example, if you wanted to paste the sequence into a web
program that requires sequences in FASTA format, you could
set the format to FASTA. For now, we'll view the complete
GenBank entry, which is the default. Click "Run" to
view. |
 |
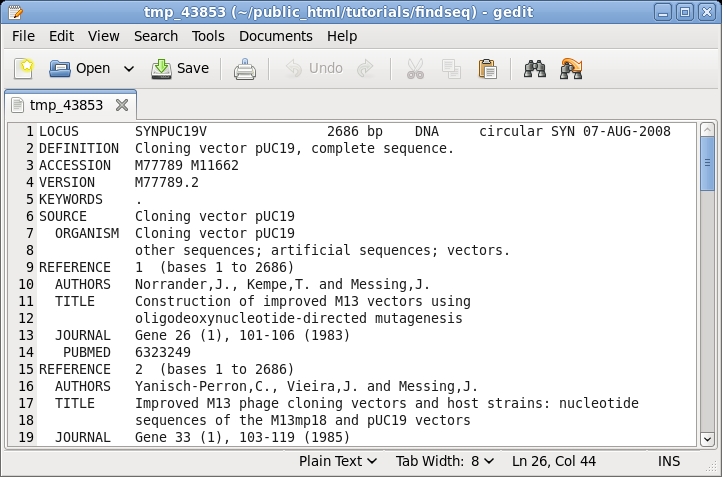
| The
GenBank file will pop up in the default text editor for
your BIRCH installation, in this case, gedit. |
 |
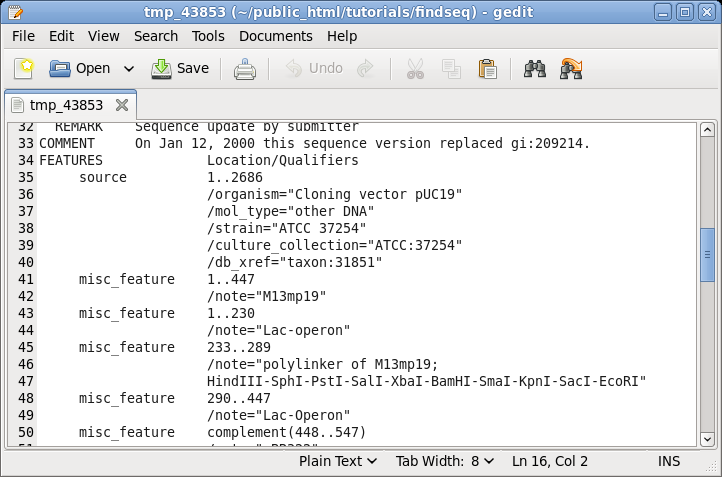
| It
is often useful to keep the sequence view open on the
screen for reference while doing other tasks. For example,
if you scroll down to the FEATURES table, you can see the
annotations for different parts of this vector. |
 |
| A
more elaborate program for viewing sequences and their
features is the Artemis Genome Browser. In bldna, choose Database
--> artemis. Artemis is a sophisticated genome browser and annotator, used in many genome projects. The wide array of functions and capabilities of artemis are beyond the scope of this tutorial. However, an introduction to Artemis is found in the BIRCH tutorial . See Genome Visualization with ARTEMIS for an in-depth introduction. |
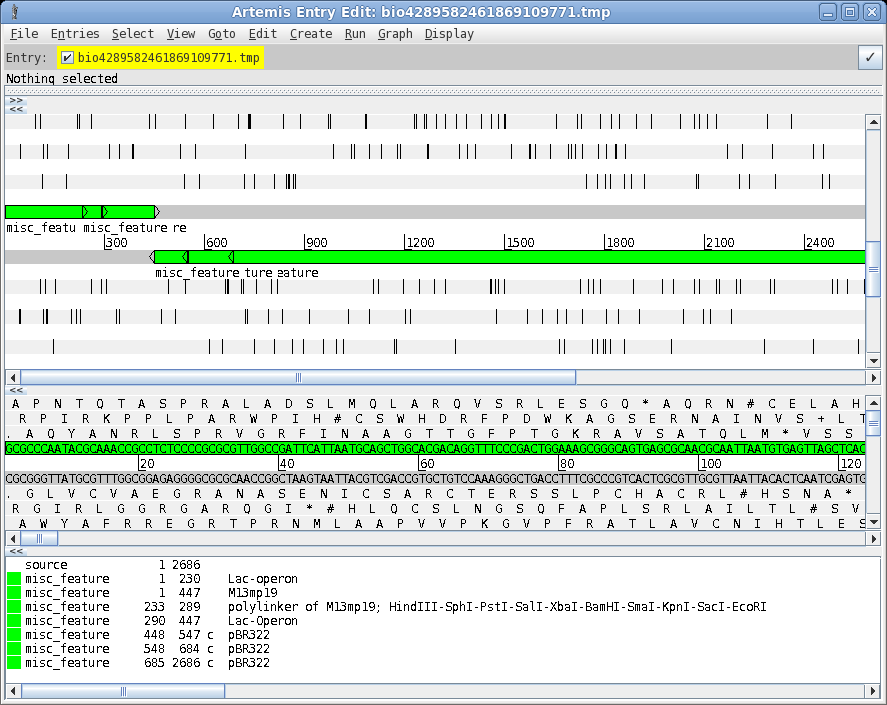 |
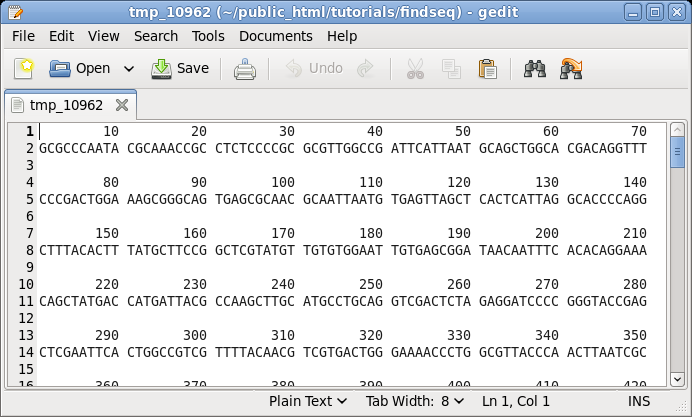
| First,
let's try printing a sequence along with its translation
in three reading frames using NUMSEQ. Choose DNA/RNA
--> NUMSEQ. A menu will pop up allowing us to set
different parameters for printing the sequence. At this
point, don't change any parameters. Just click on Run. By default, NUMSEQ will print sequences in 7 groups of 10 nucleotides per line. |
 |
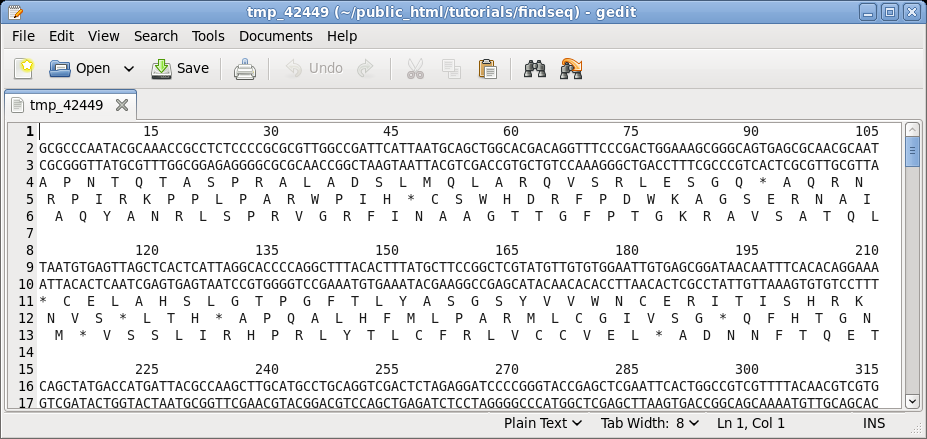
| Say
that we wanted to see both strands of the sequence, along
with a translation of the forward strand. Re-run NUMSEQ
after setting the following parameters: write in GROUPs of 15 Both strands TRANSLATION: Yes Reading frames: Three Click on Run to proceed. |
 |
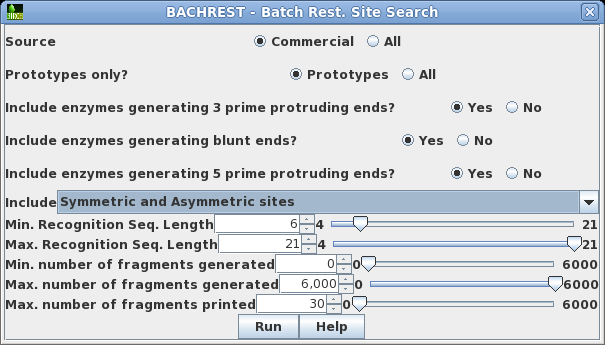
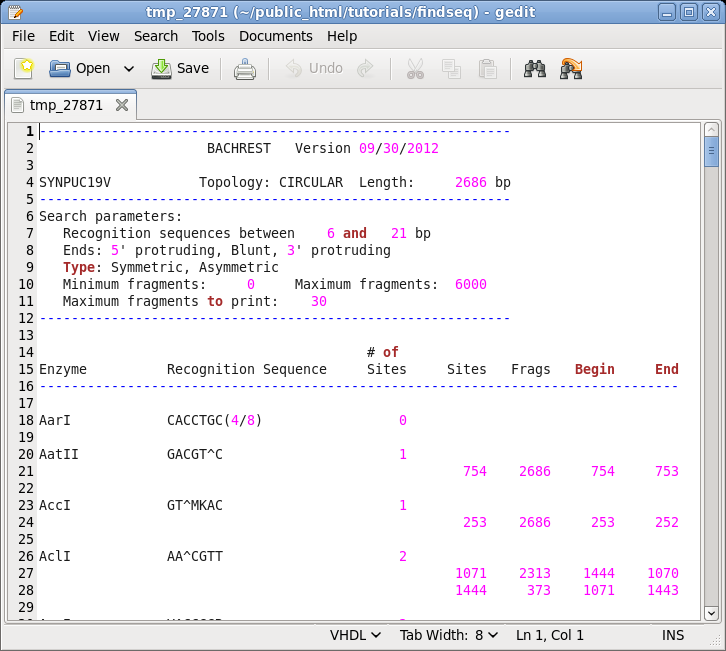
| Choose
DNA/RNA --> BACHREST. The BACHREST menu lets you customize your search base on whether or not an enzyme is comerically available, the type of ends it generates, whether or not the recognition sequence is symmetric or asymmetric, the length of the recognition sequence, or the number of fragments generated. To see the output with the default settings, click on Run. |
 |
Things
to note:
|
 |
| First,
make sure you have a fresh blncbi window. If you have
blncbi open, you can create a new window with File
--> New Window. Otherwise, launch blncbi from the
BIRCH launcher using Data Mining --> blncbi.
|
 |
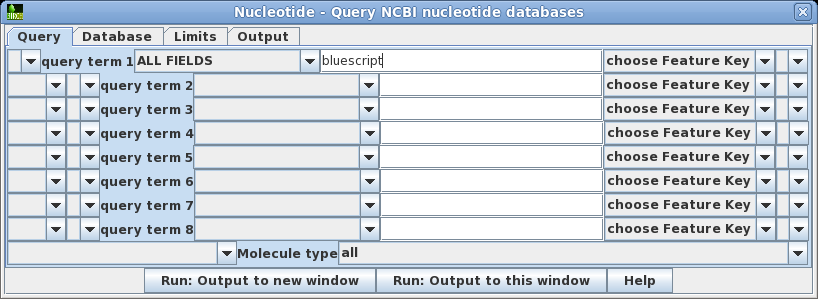
| Open
the query builder using Database -->
Nucleotide. Let's do the simplest search first. For
query term 1, the default is to search ALL FIELDS. Set the
search term to 'bluescript'. Click on Run:
Output to a new window to begin the search. |
 |
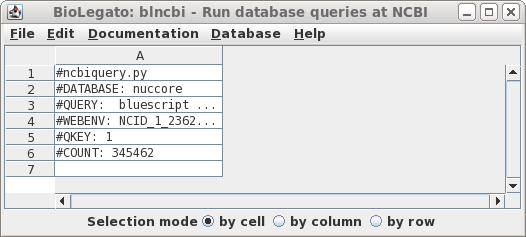
| There
are 345462 hits. In retrospect, this number shouldn't be
too surprising, because the Bluescript vectors and their
derivatives have been widely-used in cloning for decades. Let's try limiting the search by changing the search field to Title, so that only those entries in which Bluescript in which 'bluescript' appears in the title will be returned. |
 |
| The
Bluescript-related vectors are probably a very small
percentage of those hits.That means that we can eliminate
most clones by limiting the search to the GenBank
Synthetic division, which only has synthetic sequences. We
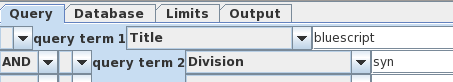
join the two search terms by choosing 'AND', and rerun the
search. |
 |
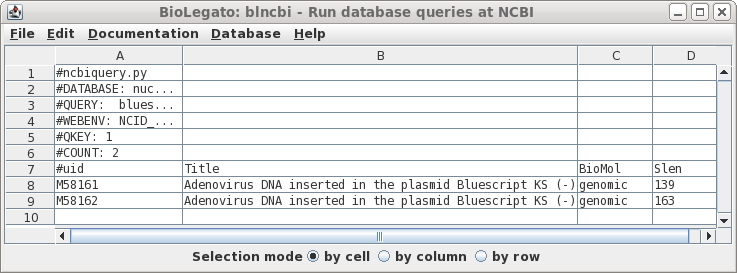
| Well
that was disappointing. This example illustrates that searches of the NCBI databases can be counterintuitive. I have no idea why the actual vectors themselves weren't found, because as we'll see later, they are in fact in the Synthetic division. (Repeating the search using variants such as "Bluescript" and "SYN" gives the same result). |
 |
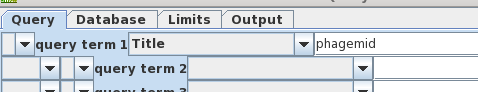
| Looking
again at bluescript.pdf, we see that the term
'phagemid' is prominent in the title. Let's us this as the
search term instead of 'bluescript'. Also, turn off
AND and remove 'syn' from query term 2, because that term
caused us to miss bluescript previously. |
 |
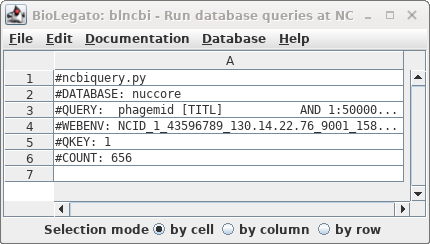
| Okay, 656 hits are a manageable number to
scan by inspection. We didn't get the actual hits because by
default blncbi will only show hits if there are 500 or
fewer. |
 |
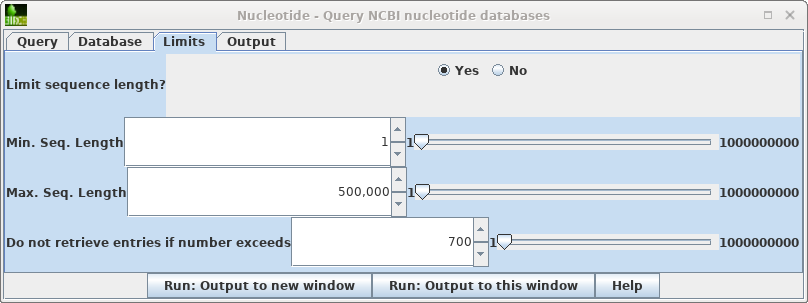
| To
see the hits go to back to the Nucleotide query and open
the Limits tab. Change "Do not retrieve if number exceeds" to something larger than 656 eg. 700. Repeat the search. |
 |
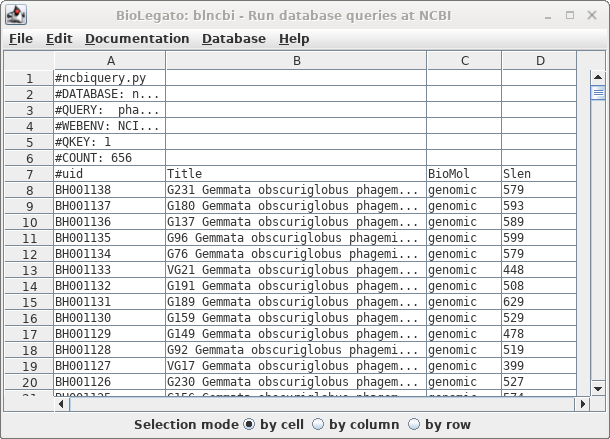
| The
result is shown at right. |
 |
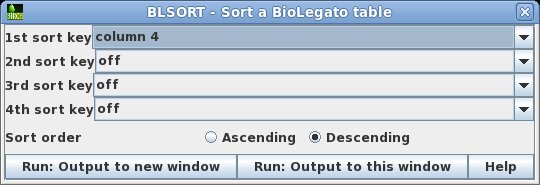
| As
a quick way to see if the Bluescript vectors are in the
list, you could try sorting the output by sequence length.
Choose Edit --> BLSORT and set
the 1st sort key to column 4 (shown as D in BioLegato). |
 |
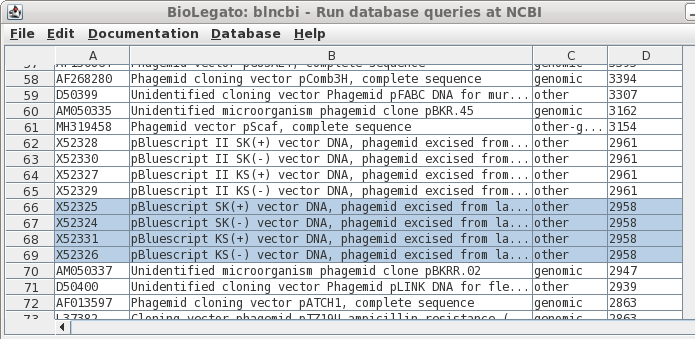
| If
you remembered that Bluescript was just a bit under 3kb in
length, you could try scrolling through the sorted output
to the correct size range, as shown in column D. Success! Select all four by holding down the CTRL key and clicking on each Accession number. |
 |
| Retrieve
these entries using Database --> SEQFETCH. |
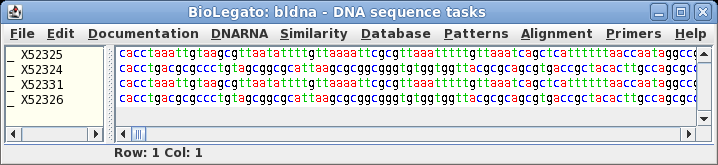 |
Select the four
sequences in bldna, and choose File --> View sequences, with
the output format set to GenBank. A quick look at the LOCUS
lines of the four sequences in this file will verify that these
sequences are indeed in the Synthetic (SYN) division.
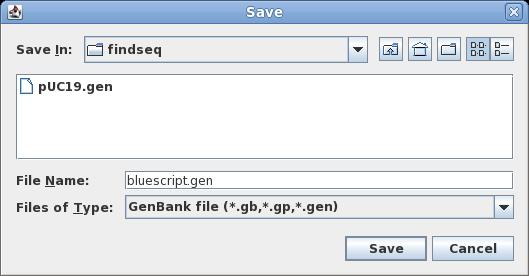
| To
save all sequences to a single file, choose File
--> Save ALL as. Set the File Name to bluescript.gen, and make sure Files of Type is set to GenBank. Save the file. |
 |