TUTORIAL:
|
Nov. 14, 2024 |
TUTORIAL:
|
Nov. 14, 2024 |
NUMSEQ documentation: $doc/fsap/numseq.txt
BACHREST documentation: $doc/fsap/rest.txt
{brassica:/home/plants/frist}cd
do next
step if $HOME/tutorials doesn't exist
{brassica:/home/plants/frist}mkdir
tutorials
create directory
for this tutorial
{brassica:/home/plants/frist}mkdir
tutorials/sequence
{brassica:/home/plants/frist}cd
$birch/tutorials/bioLegato/sequence
the location of birch
($birch) is /home/psgendb in this example
next, copy GenBank
files to new directory
{brassica:/home/psgendb/tutorials/bioLegato/sequence}cp
*.gen
$HOME/tutorials/sequence
return to $HOME directory verify that
new files and directories are
present
{brassica:/home/plants/psgendb/tutorials/bioLegato/sequence}cd
{brassica:/home/plants/frist}ls
-l
drwx------
1
frist drr 512 Oct 31
10:11 tutorials/
{brassica:/home/plants/frist}cd
tutorials
{brassica:/home/plants/frist/tutorials}ls
-l
drwx------
3
frist drr 512 Oct 31
10:11 sequence/
{brassica:/home/plants/frist/tutorials}cd
sequence
{brassica:/home/plants/frist/tutorials/sequence}ls
-l
-rw------- 1 frist
frist 5404 Oct 31 10:13 X52331.gen
-rw------- 1 frist frist
10739 Oct 31 10:13 PBI101TD.gen
-rw------- 1 frist
frist 8278 Oct 31 10:13 pBSGUS.gen
-rw------- 1 frist
frist 3674 Oct 31 10:13 PEACAB15.gen
bioLegato is a program that runs other programs. bldna
is an instance of bioLegato that contains functions and
programs specifically for DNA sequences. bldna can be
launched either from the BIRCH launcher or from the command line.
As you will see, the program runs in the window in which a
bioLegato instance was started. The bioLegato instance generates
the keystrokes that you would normally be typing.
To illustrate the point, let's try running NUMSEQ from bldna. We
will demonstrate to launch bldna from the command line and from
BIRCH.
{brassica:/home/plants/frist/tutorials/sequence}bldna

From the interface, select the
option Sequence --> bldna -
DNA sequence analysis system.
This will open another window for bldna.
| IMPORTANT NOTES: 1.While a bioLegato instance is running, the terminal window can not be used for other commands. If you need to type commands, open another terminal window. Alternatively, you can launch bldna (or any program) in the background by including the ampersand character at the end of the command eg. bldna & . The window would now be free to use for other commands. 2. Although bioLegato can read files from any directory, it's best to launch a bioLegato instance from the directory in which you plan to work. |
Read in PEACAB15.gen:
File --> Open |
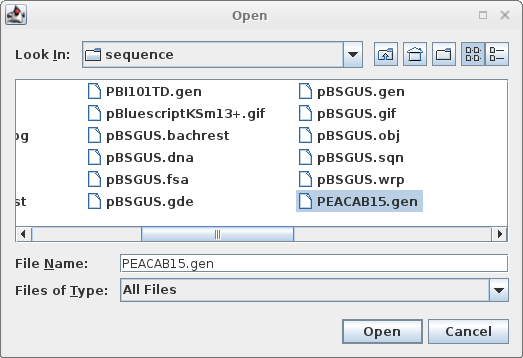 |
Click on the filename, and click 'Open'. |
 |
| Hint: There
are 2 steps to running a program from a bioLegato
instance 1. Select sequence(s) - either:
2. Choose a program from one of the menus
|
|
To run numseq, click on PEACAB15 and choose DNARNA --> NUMSEQ. The numseq menu appears, containing menu items for all parameters in the NUMSEQ Parameters menu. |
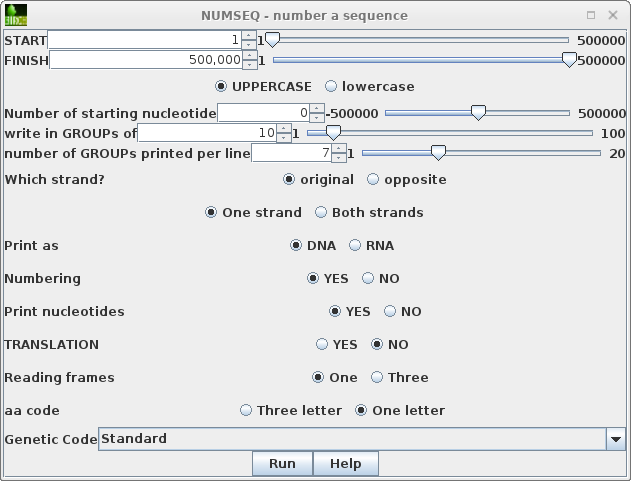 |
HINTS ON bioLegato MENUS:
|
|
When you click OK, bldna saves the specified sequence in a temporary file, and runs numseq. Numseq reads in the temproary sequence file and prints it out according to the parameters sent to it by bldna. Output is stored in a temporary file, which is opened in a text editor. Normally, the temporary output file (eg. bioLegato2155281324855234117.tmp.out) will be deleted when you quit the Text Editor window. To save the file, choose File --> Save As and type in a name for the output. It's a good idea to include a .numseq file extension to indicate that this file is output from numseq. |
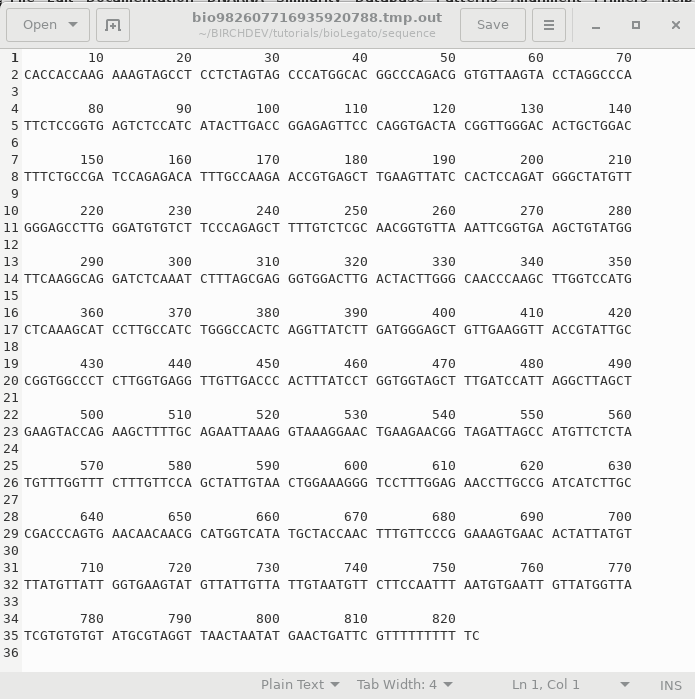 |
Because the output is ASCII text, you can do lots of things with
it, including importing it into a word processor, pasting it into
another window, mailing it, or even using it as input for other
sequence programs. In the latter case, the output will probably
need to be modified to conform to the desired input file format
eg. Pearson/Fasta.
To view both strands:
set "Both strands"
To translate in 3 reading frames:
TRANSLATION: Yes
Reading frames: Three
write in GROUPs of: 15
number of GROUPs printed per line:
NUMSEQ breaks up the sequence into groups of nucleotides, numbering each group. For translation, GROUP must be divisible by 3, because translation is done in discrete codons of 3 bases each. Because the resultant output line will be 15 groups of nucleotides times 7 groups per line (ie. 105 characters per line), you will have to stretch the output window to prevent text wrapping.
To limit printing to only part of the sequence eg. bases 200 - 400:
START: 200
FINISH: 400
TRANSLATION: No
To view the opposite strand of the same region:
Which strand: opposite
START: 400
FINISH: 200
This example illustrates that creating an opposite strand requires two steps. First, we have to specify the strand as 'o' (opposite) rather than 'i' (input strand). This causes the bases to be complemented. However, if all we do is complement the input strand, then the opposite strand would be printed 3' to 5', because we would be starting at 200 and ending at 400. Therefore, START must be set to 400, and FINISH to 200.
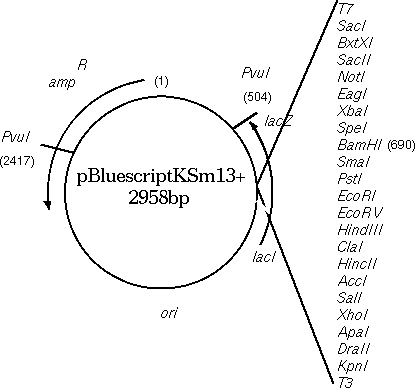 Circular DNA molecules
require a bit of thought. Since printing is always done 5' -->
3', the direction (clockwise vs. counterclockwise) determines the
strand, or vice versa. Consider the Bluescript cloning vector
(GenBank X52331). Conceptually, one base must be arbitrarily
labeled as 1. In the GenBank entry, 1 is the first base in the
file, and 2958 is the last base in the file. In the physical
plasmid, of course, base 2958 is adjacent to 1.
Circular DNA molecules
require a bit of thought. Since printing is always done 5' -->
3', the direction (clockwise vs. counterclockwise) determines the
strand, or vice versa. Consider the Bluescript cloning vector
(GenBank X52331). Conceptually, one base must be arbitrarily
labeled as 1. In the GenBank entry, 1 is the first base in the
file, and 2958 is the last base in the file. In the physical
plasmid, of course, base 2958 is adjacent to 1.
In NUMSEQ, the START, FINISH and WHICH parameters govern which parts of the sequence are displayed.
To view the top strand of the PvuI (CGAT^CG) fragment going clockwise from 2417 to 503:
1) START: 2417
2) FINISH: 503
7) WHICH: Original
Since you're only considering 1 strand at a time, you want to start with 2417, which is the 5' end of the small PvuI fragment, on the original strand.
To print the same sequence on the other strand, we can't just change WHICH to 'Opposite".
1) START: 2417
2) FINISH: 503
7) WHICH: Opposite
Try it and you'll see that what you get is the large PvuI fragment going from 2417 to 503, and that this fragment doesn't even terminate where PvuI would cut. It's best to visualize the fragment ends as illustrated below:
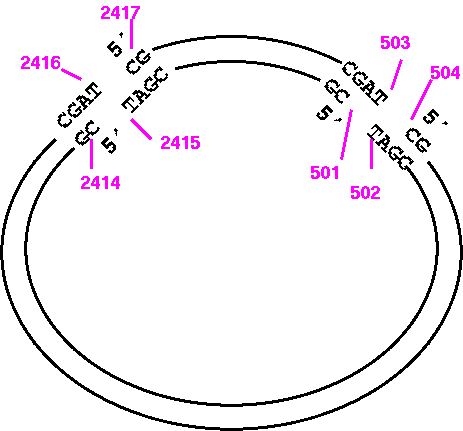
So the correct way to print the opposite strand of the small fragment would be:
1) START: 501
2) FINISH: 2415
7) WHICH: Opposite
Example: Simulated restriction digest of a
pBluescriptKSm13+ at BamH1
The BamHI site is at 690 on the input strand, meaning that the 5'
end of the BamH1 site on the original strand is at position 690.
Thus:
1) START: 690
2) FINISH: 689
7) WHICH: Original
If we wanted the inverse complement (ie. counter clockwise), the NUMSEQ parameters would be
1) START: 693
2) FINISH: 694
7) WHICH: Opposite