| Warning! Most multiple alignment programs will align either DNA or amino acid sequences. However, it's important to know that unless nucleic acid sequences are very closely-related, with few gaps (eg. tRNA, rRNA genes), a reliable multiple alignment (or for that matter, even a pairwise alignment) is almost impossible. The reason is that nucleic acids use a 4-letter alphabet, allowing many equally good alignments form a set of sequences. In contrast, the 20-letter amino acid alphabet drastically decreases the number of possible alignments, so that an obvious 'correct' alignment is usually possible to find. |
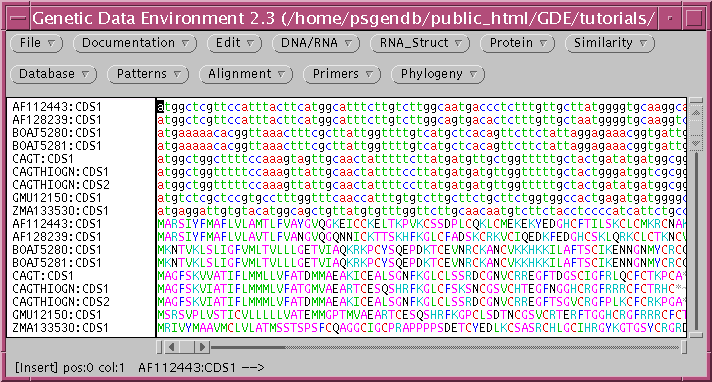
Select all amino acid sequences an open the Alignment menu, which shows programs related to multiple sequence alignment
Choose TCOFFEE, which brings up the following menu:

Since the number of sequences is small, we have the luxury of doing several of the steps using more time-consuming but more thorough methods. First, choose the dynamic programming method of Myers and Miller, rather than FASTA, which will should give optimal pairwise alignments. As well, compute the dendrogram using the extended library.
TCOFFEE sends the alignment to a new GDE window.
:

 Warning!: Do not use guide trees
generated by clustal or other multiple alignment programs for any
purpose eg. phylogenetic analysis of sequence or species
evolution. These trees are based on pairwise alignments, and
therefore do not contain the evolutionary information found in the gaps
that are present in the completed alignment. Once you have an
alignment, you can then go back and construct a phylogenetic tree. Warning!: Do not use guide trees
generated by clustal or other multiple alignment programs for any
purpose eg. phylogenetic analysis of sequence or species
evolution. These trees are based on pairwise alignments, and
therefore do not contain the evolutionary information found in the gaps
that are present in the completed alignment. Once you have an
alignment, you can then go back and construct a phylogenetic tree. |

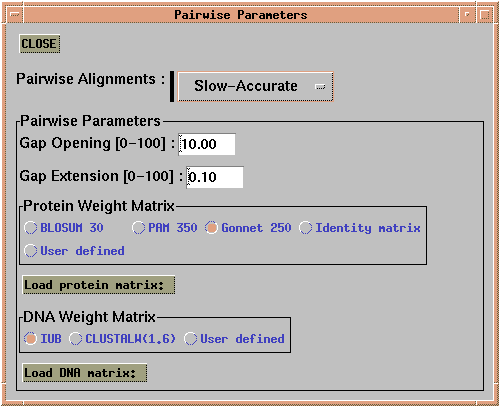
The first step in a multiple alignment using CLUSTALX is to perform all pairwise alignments between sequences. Choose 'Alignment --> Alignment Parameters --> Pairwise Alignment Parameters'. This menu lets you choose gap penalties and scoring matrices.
Next, 'Alignment --> Alignment Parameters --> Multiple
Alignment Parameters' allows the choice of similar parameters for
building the multiple alignment from the pairwise alignments. Although
the default parameters are the same, it is possible that parameters
might be adjusted at this stage for more closely-related sequences,
since adjacent sequences in an alignment are more likely to be
closely-related to each other.

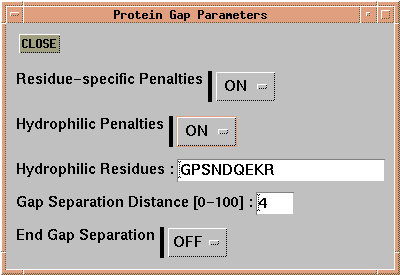
Finally, for amino acid sequences, 'Alignment --> Alignment
Parameters --> Protein Gap Parameters' allows the setting of gap
penalties dependent on structural or chemical criteria.
To begin the alignment, choose 'Alignment --> Do Complete Alignment'.
MRTRANS by Bill Pearson aligns DNA sequences using the corresponding
amino acid alignment as a guide. Thus, if you have aligned a set of
amino acid sequences, it is straightforward to generate the
corresponding DNA alignment. MRTRANS requires two files for input, in
Pearson/FASTA format. The first file contains the unaligned protein
coding sequences, and the second file contains the corresponding amino
acid sequences, aligned by a program such as TCOFFEE or CLUSTALX.
| Notes: 1) MRTRANS needs the DNA and aligned amino acid sequences to have the same names. During the extraction process, during translation, the names may be modified, so it may be necessary to change names in 'File --> Get Info', before you export to a .wrp file 2) Where two or more copies of a gene are present in a single entry (eg. CAGTHIOGN:CDS1 and CAGTHIOGN:CDS2), it is necessary to give them each unique names so that MRTRANS can distinguish them. Since the CDS extensions will be removed when MRTRANS is run, one solution is to delete CDS but retain the number (eg CAGTHIOGN_1 and CAGTHIOGN_2). The current GDE implementation of TCOFFEE and CLUSTALX usually handle these steps automatically. Nonetheless, if you are having problems with mrtrans, make sure that the names of sequences are the same for both protein and nucleic acid sequences. |
Continuing with our earlier example, select all of the
defensin DNA sequences,

and choose 'File --> Export Foreign Format' .
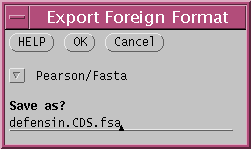
Save the sequences in FASTA format, to the file defensin.CDS.fsa.
Next select the amino acid alignment. Note that only the actual
amino acid sequences, and NOT the PIMA-generated patterns, are selected
for export.

and choose 'File --> Export foreign format' to save as defensin.tcoffee.fsa.

To run MRTRANS, choose 'Alignment --> MRTRANS', and type in the
DNA and protein alignment filenames:
![]()
The aligned DNA sequences appear in a new GDE window:
![]()
| Hint: One of the most common errors is
to switch the names of the Multiply-Aligned Protein File and hte Unaligned
DNA file, which, of course, will fail. |
Alignments can be edited directly in GDE. By default, only gaps can be edited out, although it is possible to delete amino acids or nucleotides if protections are changed (File --> Protections).
Sometimes, it is useful to set several sequences to act as a group.
For example, if we wanted to make the second group of Cys residues in
the bottom three sequences line up with the others, we could select
BOAJ5280, BOAJ5281 and ZMA133530, and choose 'Edit --> Group' The
'1'
at the left of these sequences indicates that changes made to any of
these sequences will be made to all. Thus, deletion of a gap character
could be done by clicking on the C in the bottom sequence (CSAS) and
pressing the "Delete" key. To restore the downstream part of the
alignment, another gap will have to be inserted elsewhere.

See $doc/GDE/GDE2.2_manual.pdf
for a more in depth description of how to edit alignments.
In many cases you need an alignment displayed as editable text. This might be true if you wanted to be able to import the alignment into a word-processor or HTML editor for further modification, such as coloring or underlining certain characters. Choosing 'Alignment --> REFORM' will print out an alignment in which amino acids matching the consensus are indicated by dots:
10 20 30 40 50 60 70
Maxxxkxxaxxflmxtlx-------xaxx-----xxxxxxxcexxsxxfkgxCxsxrxcxxvcxxxek--
CAGT:CDS1 ..gfs.vv.ti...ml.v.......f.td.....mmaeaki..al.gn...l.l.s.d.gn..-rr.g..
CAGTHIOGN_..gfs.vi.ti...mm.v.......f.td.....mmaeaki..al.gn...l.l.s.d.gn..-rr.g..
CAGTHIOGN_..gfs.vi.ti...mm.v.......f.tg.....mvaeart..sq.hr...l.f.ksn.gs..-ht.g..
GMU12150:C.srsvplvsticvlll.l.......v.temmgptmvaeart..sq.hr...p.l.dtn.gs..-rt.r..
AF112443:C..rsiyfm.flv.am..f.......v.yg.....vqgkeic.keltkpv.--.s.dpl.qkl.mek....
AF128239:C..rsiyfm.flv.av..f.......v.ng.....vqgqnni.ktt.kh...l.fadsk.rk..iqed...
BOAJ5280:C.kntv.lslig.v.l.vl.......llgetviaqkrkpcysq.p---dkt--.evn.ckancvkkhk.il
BOAJ5281:C.kntv.lslig.v.l.vl.......llgetviaqkrkpcysq.p---dkt--.evn.ckancvkkhk.il
ZMA133530:.rivymaavmclvla.msstspsfcq.ggcigcprappppsd.tcyedl.--.sas.chlgcihrgy...
Various features of the output can be changed in the REFORM menu For
example, to print ALL amino acids at every position, the REFORM menu
would be set as follows: 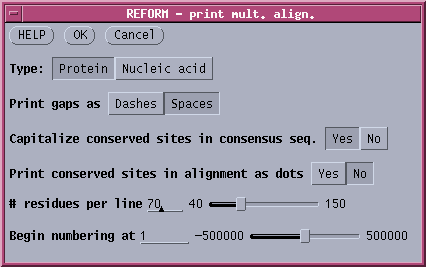
10 20 30 40 50 60 70
Maxxxkxxaxxflmxtlx xaxx xxxxxxxcexxsxxfkgxCxsxrxcxxvcxxxek
CAGT:CDS1 magfskvvatiflmmllv fatd mmaeakicealsgnfkglclssrdcgnvc rreg
CAGTHIOGN_magfskviatiflmmmlv fatd mmaeakicealsgnfkglclssrdcgnvc rreg
CAGTHIOGN_magfskviatiflmmmlv fatg mvaeartcesqshrfkglcfsksncgsvc hteg
GMU12150:Cmsrsvplvsticvlllll vatemmgptmvaeartcesqshrfkgpclsdtncgsvc rter
AF112443:Cmarsiyfmaflvlamtlf vayg vqgkeicckeltkpvk cssdplcqklcmekek
AF128239:Cmarsiyfmaflvlavtlf vang vqgqnnickttskhfkglcfadskcrkvciqedk
BOAJ5280:Cmkntvklsligfvmltvl llgetviaqkrkpcysqep dkt cevnrckancvkkhkkil
BOAJ5281:Cmkntvklsligfvmltvl llgetviaqkrkpcysqep dkt cevnrckancvkkhkkil
ZMA133530:mrivymaavmclvlatmsstspsfcqaggcigcprappppsdetcyedlk csasrchlgcihrgyk
b) JALVIEW - Graphic display and
alignment
Jalview is a feature-rich sequence alignment viewer. It can be
launched by selecting an alignment in GDE, and choosing 'Alignment
--> Jalview'.

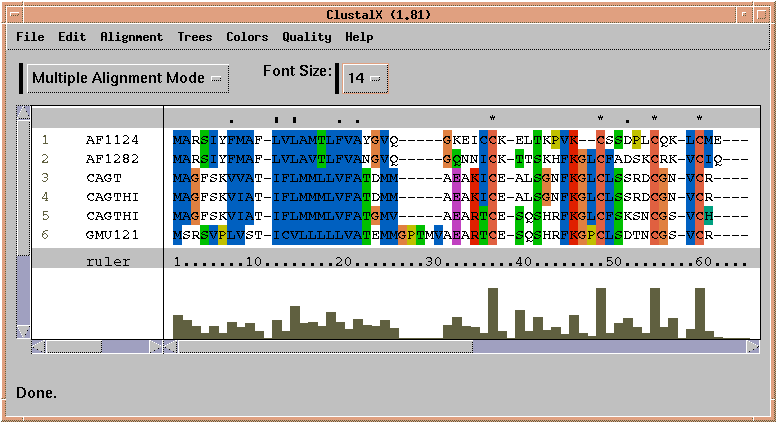
The alignment above is shown using one of several color schemes available. The Hydrophobicity color scheme shows hydrophobic residues in red, hydrophilic residues in blue, and residues of intermediate hydrophobicity in varying shades of purple.
The alignment can be written in paginated form, suitable for printing, by saving to a PostScript file. PostScript is an almost universal printer language understood by virtually all laser printers. An example of PostScript output can be seen in defensin.jalview.ps. Clicking on this link should launch ghostview or a similar PostScript viewer, which can print the file. The file could also be saved and printed to any laser printer (eg. lpr defensin.jalview.ps).
If your browser is not configured to launch a PostScript viewer, you can save the file, and convert it to PDF. Most Unix systems have the ps2pdf command:
ps2pdf defensin.jalview.ps
would create a file called defensin.jalview.pdf. Clicking on this link should launch a PDF viewer such as Adobe Acrobat or ggv.
Jalview can also do complete alignments from unaligned sequences.
For a full description of the capabilities of Jalview, see $doc/jalview/contents.html.
| Note to VNC users: Jalview, like many Java applications, has color usage issues. For example, running vncserver at 16-bit color depth (eg vncserver -depth 16) will cause the alignment window to appear completely black. Running vncserver at the default color depth of 8 seems to work (eg vncserver). |