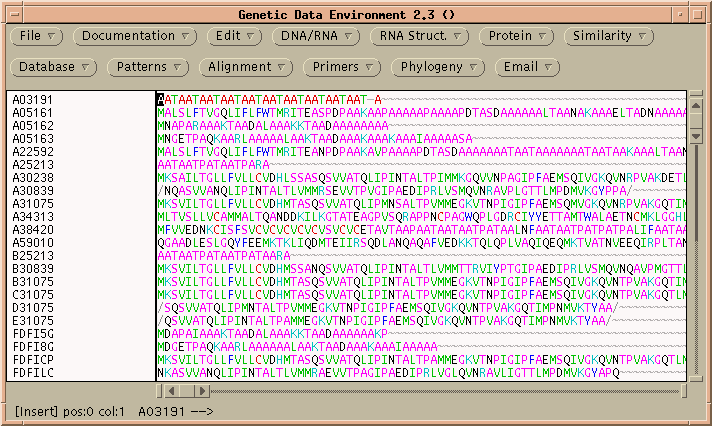
ALIGN performs rigorous global alignments. That is, all of both proteins is fit into the alignment. Choose Similarity --> ALIGN to get the ALIGN menu:
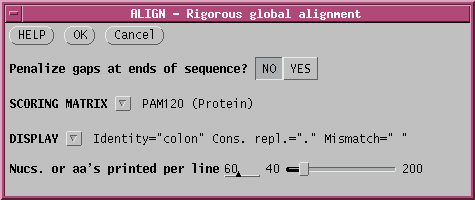
Since these sequences are known to be very closely-related, it probably makes sense to use a menu like PAM120, rather than the default, for the most precise score.
ALIGN calculates a global alignment of two sequences
version 2.0uPlease cite: Myers and Miller, CABIOS (1989)
>A30238 88 bp 88 aa vs.
>A31075 87 bp 87 aa
scoring matrix: /home/psgendb/dat/fasta/pam120.mat, gap penalties: -12/-4
80.7% identity; Global alignment score: 372
10 20 30 40 50 60
gde208 MKSAILTGLLFVLLCVDHLSSASQSVVATQLIPINTALTPIMMKGQVVNPAGIPFAEMSQ
:::.::::::::::::::.. ::::::::::::.:.::::.:: :.:.:: :::::::::
A31075 MKSVILTGLLFVLLCVDHMT-ASQSVVATQLIPMNSALTPVMMEGKVTNPIGIPFAEMSQ
10 20 30 40 50
70 80
gde208 IVGKQVNRPVAKDETLMPNMVKTYRAAK
.:::::::::::..:.:::::::: :.:
A31075 MVGKQVNRPVAKGQTIMPNMVKTYAAGK
60 70 80
(Note on output: Two windows will pop up, the output from the
search and a narrow window with names of sequences in the search
file.The
latter can usually be discarded.)
When similarity is this high, it is easier to view the mismatches, rather than the matches:
ALIGN calculates a global alignment of two sequences
version 2.0uPlease cite: Myers and Miller, CABIOS (1989)
>A30238 88 bp 88 aa vs.
>A31075 87 bp 87 aa
scoring matrix: /home/psgendb/dat/fasta/pam120.mat, gap penalties: -12/-4
80.7% identity; Global alignment score: 372
10 20 30 40 50 60
gde208 MKSAILTGLLFVLLCVDHLSSASQSVVATQLIPINTALTPIMMKGQVVNPAGIPFAEMSQ
x xx x x x X x x X
A31075 MKSVILTGLLFVLLCVDHMT-ASQSVVATQLIPMNSALTPVMMEGKVTNPIGIPFAEMSQ
10 20 30 40 50
70 80
gde208 IVGKQVNRPVAKDETLMPNMVKTYRAAK
x xx x X x
A31075 MVGKQVNRPVAKGQTIMPNMVKTYAAGK
60 70 80
The ALIGN output also shows substantial divergence, compared to that seen for among the two proteins.
ALIGN calculates a global alignment of two sequences
version 2.0uPlease cite: Myers and Miller, CABIOS (1989)
>A31075 87 bp 87 aa vs.
>B30839 91 bp 91 aa
scoring matrix: BLOSUM50, gap penalties: -12/-2
62.6% identity; Global alignment score: 330
10 20 30 40 50
gde208 MKSVILTGLLFVLLCVDHMT-ASQSVVATQLIPMNSALTPVMMEGKVTNPIGIPFAEMSQ
:::::::::::::::::::. :.::::::::::.:.::: ::: .: : ::: .. .
B30839 MKSVILTGLLFVLLCVDHMSSANQSVVATQLIPINTALTLVMMTTRVIYPTGIPAEDIPR
10 20 30 40 50 60
60 70 80
gde208 MVGKQVNRPVAKGQTIMPNMVKTYA--AGK-
.:. :::. : : :.::.::: : : :
B30839 LVSMQVNQAVPMGTTLMPDMVKFYCLCAPKN
70 80 90
For sequences that are not closely-related, it is not appropriate to use global alignment programs such as ALIGN, because a global alignment tries to force all parts of both sequences into an alignment, regardless of whether those parts share significant similarity. LALIGN and LFASTA search for local alignments, building alignments locally until extension of the alignment results in a decreased, rather than an in increased score.
Select T44768 and T51008 and choose Similarity --> LALIGN.
The
LALIGN menu will pop up. Since we're assuming distant relationships,
choose
the BLOSUM50 scoring matrix. The output gives several alignments, the
longest
shown below: