TUTORIAL: PHYLOGENETIC ANALYSIS USING
DISTANCE
METHODS
PHYLIP Main documentation: $doc/Phylip/main.html
PHYLIP Distance methods: $doc/Phylip/distance.html
PHYLIP DNADIST: $doc/Phylip/dnadist.html
PHYLIP PROTDIST: $doc/Phylip/protdist.html
PHYLIP FITCH: $doc/Phylip/fitch.html
PHYLIP PROTDIST: $doc/Phylip/kitsch.html
PHYLIP NEIGHBOR: $doc/Phylip/neighbor.html
ATV tree viewer: $doc/atv/atv_documentation.pdf
The PHYLIP programs are command line
programs, but can
be run by GDE
The programs in the PHYLIP package are
interactive programs
designed to be run at the command line. GDE can run these programs by
generating
the keystrokes needed to set programs parameters.
Construction of a phylogeny using distance methods
involves two steps:
- GDE runs DNADIST or PROTDIST, to construct a distance
matrix.
- The distance matrix is used to construct a
phylogenetic tree, using any of a number of methods implemented in the
programs FITCH, KITSCH or NEIGHBOR.
|
Example: Plant Type III Chitinases
The chitinases in plants are hydrolytic enzymes that degrade chitins
(N-acetyl
glucosamine). Although chitin does not occur in plants, in many
fungi,
it is a major component of the fungal cell wall. Not surprisingly,
chitinases
are produced in plants in response to fungi. Chitinases have been
demonstrated
to play an important role in plant defense responses. There are six
classes
of chitinases so far identified. Most known chitinases fall into the
Type
I and Type II classes. This exercise will work with a smaller class of
genes encoding Type III chitinases.
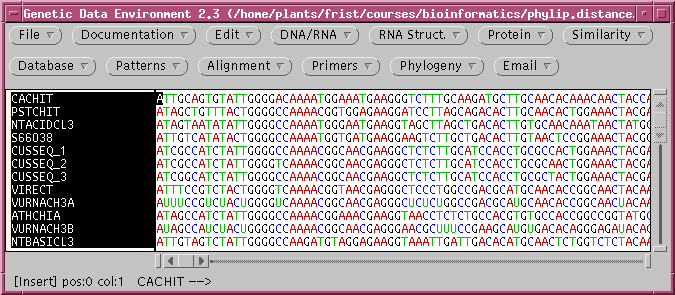
1. The dataset
The file chitIII.mrtrans.gde is a
GDE
format file containing protein coding sequences (CDS) from chitinase
III
genes. These DNA sequences have already been aligned using Pearson's
mrtrans
program, which reads a set of unaligned DNA sequences and aligns them
accroding
to a set of aligned proteins.
Create a directory called distance, and save
chitIII.mrtrans.gde
to this directory. Open the file in GDE:
2. A quick phylogeny using FITCH
For routine distance tree construction, the
method of Fitch and Margoliash is the method of choice. FITCH allows
for variable rates of evolution indifferent lineages, and iterates the
tree to minimize the least squares distance across the entire tree.
Although Neighbor-Joining is faster, it is also much less thorough,
considering one one tree. It is probably the least
rigorous
method for constructing a phylogeny . To run FITCH, choose Phylogeny
--> DNA Distance Methods. Fitch-Margoliashs is the default
method.
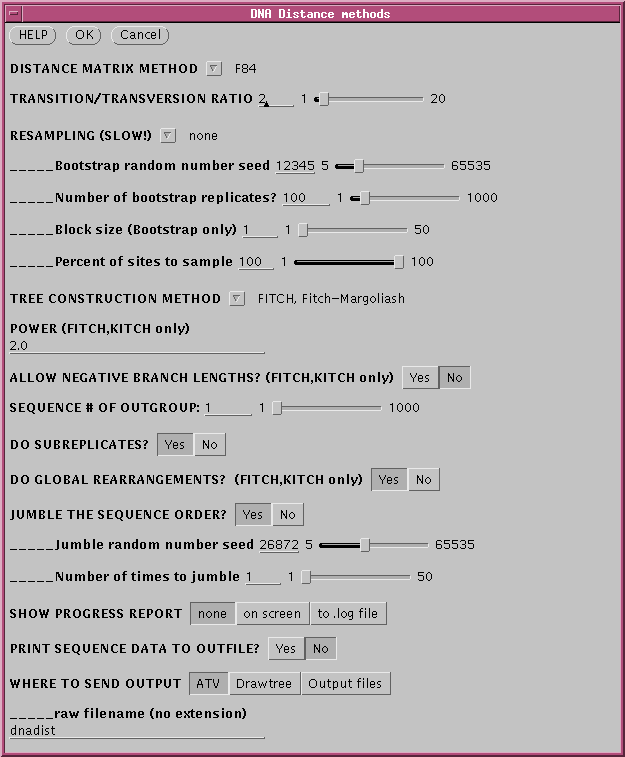
Since all distance method are
sensitive to the order in which sequences are added to the tree, set a
random number seed for jumbling the sequence order.
DNADIST will calculate a distance matrix, and then FITCH will
run,
and by default, 3 windows will appear.
OUTFILE -
the report on the phylogeny
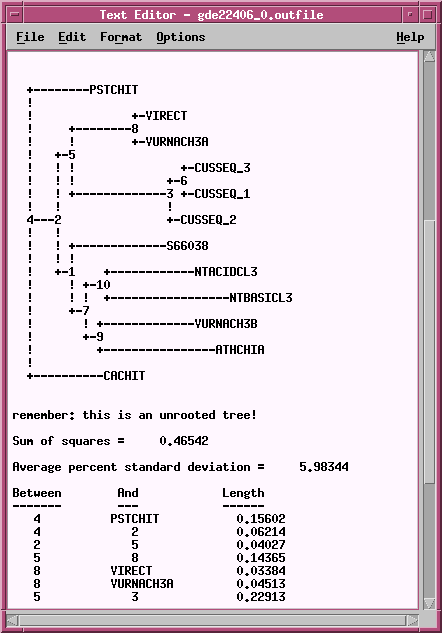
TREEFILE -
the machine -readable treefile. Readable
by
programs such as DRAWTREE, DRAWGRAM, and ATV.
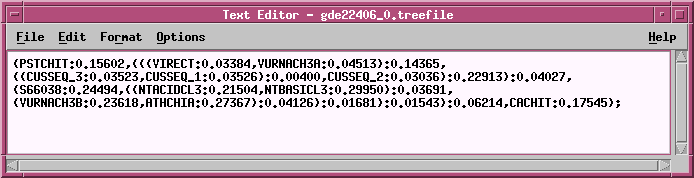
TREEFILE - the treefile in the ATV tree editor.
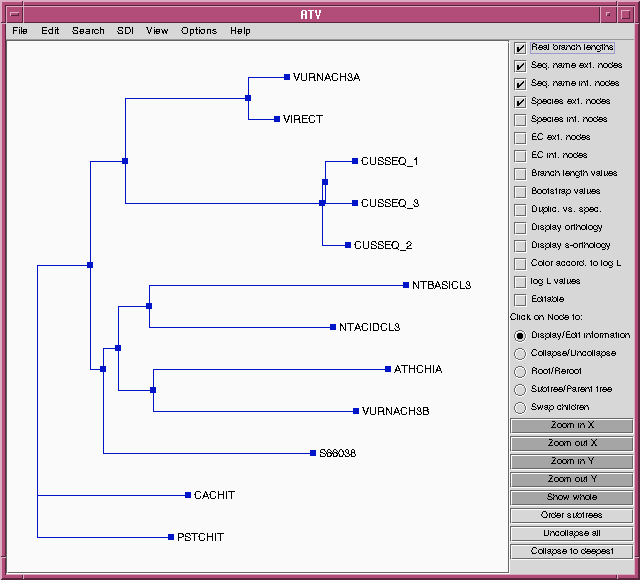
| Hint: Each of these files need to be saved separately, if you
wish
to save them. Give them all the same base name, but different
extensions,
such as chitIII.dna.fitch.outfile, chitIII.dna.fitch.treefile.
Note: Do NOT save the contents of the ATV window using
the .treefile
extension. You will overwrite the original treefile. ATV can save files
in NHX
(New Hampshire, extended) format, which will preserve any
changes made in ATV. In most cases,
you
can just save the .treefile and read it into ATV, treetool, or
other tree drawing programs whenever
you
want to work with it.
|
3. Phylogeny using amino acid sequences.
Since also possible to construct distance matricies for multiple
alignments of amino acid sequences, the same programs (FITCH, KITSCH,
and NEIGHBOR) can be used to construct distance trees. The file chitIII.pro.tcoffee.gde contains
the chitinase III proteins aligned using TCOFFEE.
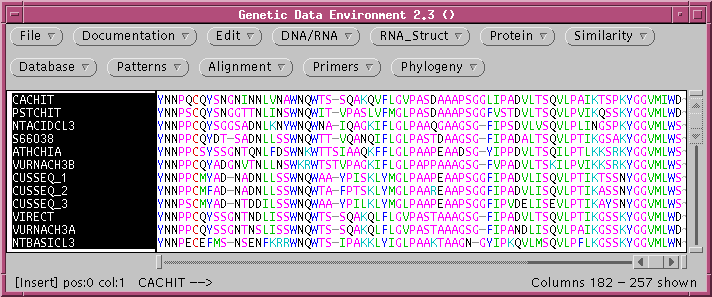
Some of the parameters for construction of the distance matrix using
PROTDIST are different from those for DNADIST. These include
several different methods for constructing distance matrices, as well
as a choice of alternative genetic codes, where appropriate.
Once the distance matrix is constructed, there is no difference in
computation of the phylogenetic tree, so all parameters are the same as
previously.
FITCH will produce an outfile (chitIII.pro.fitch.outfile)
and a treefile (chitIII.pro.fitch.treefile)
similar to those with the DNA alignment. For comparison, the
treefile is shown in ATV below:
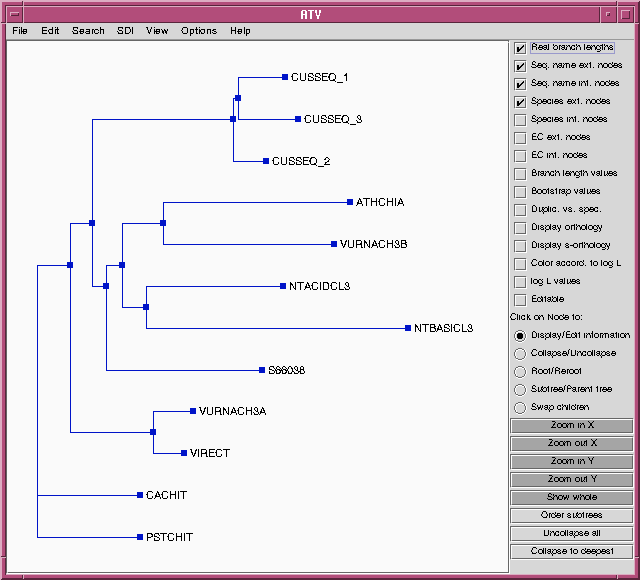
While this may look like a different tree than that produced using the
DNA alignment, the topologies (ie. the order of branching) are
identical. To prove this, we can choose the "Swap children" option in
ATV, and then click on internal nodes to rotate the branches.
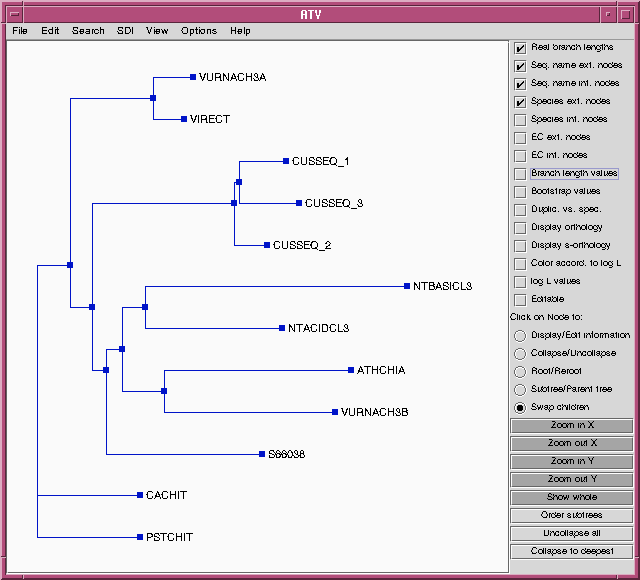
Comparison with the tree from the DNA alignment shows that these
trees have identical topologies, and similar lengths for most
branches.