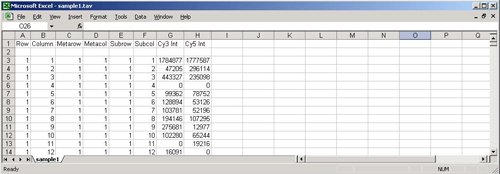
A TAV file containing only the required fields
The original TAV (TIGR ArrayViewer) file type was an eight-column, tab-delimited text format developed at TIGR for the purposes of storing the intensity values of the spots on a single slide. It is written out by the program TIGR Spotfinder and contains one row for each spot. The first six columns of the file contain positional data for the spots and are followed by two columns of intensity data.
These eight columns are required by MeV for display and analysis of experimental data. Optional columns can contain flags, annotation, Genbank numbers, etc. It is the variability of .tav files caused by these optional columns that make Preferences files necessary. Optional columns can be used to sort the spots in the , by choosing the appropriate column from the menu.
A flag is simply a letter code corresponding to a description of the spot:
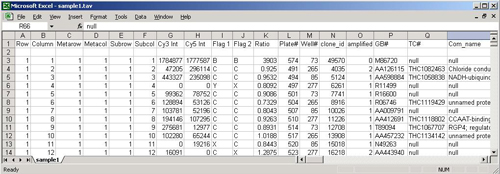
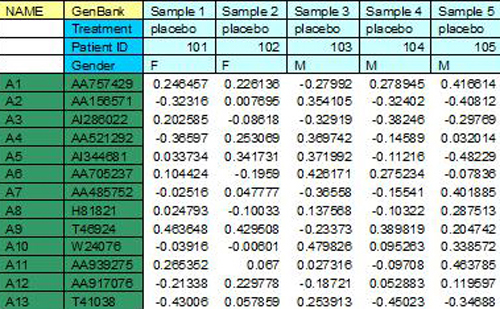
TDMS files encapsulate the expression data from multiple samples into a single tab delimited file. Each sample represented in the file will have a single dedicated column that contains the expression data for that sample. Each row, below the header rows, represents information relating to a particular spot on the slide.
The following sections describe the format of the file in detail. The image following the description contains color coded sections that relate to each of the distinct areas of the file.
(yellow, light blue, and cyan, top 4 rows in the example)
TDMS files must contain one or more header rows. These header rows must be at the top of the file. The first header row is used primarily to contain the default sample name for each sample contained in the file. The first header row also contains descriptive gene annotation field names (yellow) over the columns dedicated to gene annotation. Additional header rows may be present and each additional header row contains additional sample annotation.
(green (annotation), yellow (annotation field names)
The TDMS format permits any number of annotation columns, which must occupy the left most columns of data in the file. Each of these annotation columns contains annotation corresponding to the spot represented by that row of the file. Each of the annotation columns has a label in the top header row to indicate the annotation type.
(uncolored section with numeric data)
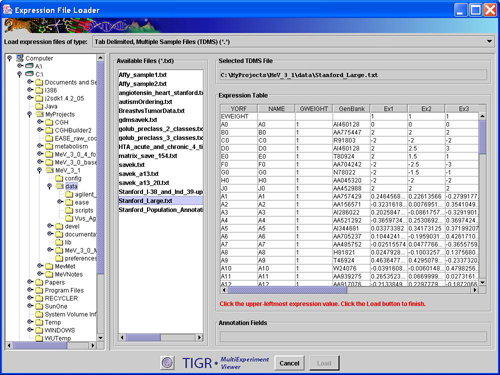
The Expression data is arranged such that there is one column for each sample represented in the file. The position of the expression data column for a particular sample is beneath the header’s sample label for that sample. See the TDMS file loader figure below as an example file displayed within the preview window.
Microarray data in the Genepix file format can be easily converted to tav file format using TIGR’s Genepix Converter application. This program is freely available from the TIGR website as part of the MADAM package). The latest version of the converter can convert multiple files in batch mode. See the help files for the Genepix Converter for more details. Files converted when the “Keep all Genepix data” box is checked will be extremely large and may cause MeV to run slowly. Leaving this box unchecked will cause only the data required by MeV to be written to the output tav file.
A MultiExperimentViewer or .mev file is a tab-delimited text file that contains coordinate and expression data for a single microarray experiment. A single header row is required to precede the expression data in order to identify the columns below. With the exception of optional comment lines, each remaining row of the file stores data for a particular spot/feature on the array.
MeV and other TM4 software tools will consider comment lines non-computational. A comment line must start with the pound symbol ‘#’, and can be included anywhere in the file. If the pound symbol is the first character on a line, the entire line (up to the newline character ‘\n’) will be ignored by the software tool.
.mev files will typically contain at least one comment at the top of the file with the following information. This information is optional. The format and fields contained within these comments are subject to change.
: Version number based on revisions of expression data
: The version of the .mev file format document
: Date of file creation or update
: Owner or the person responsible for creating the file
: id from the analysis table that corresponds to this set of expression values
: slide_type that this array is based on
: Number of rows of expression (eg. non-header)data in input files
:Number of rows of expression (eg. non-header) data in this file
: Software tool used to create the file
: Common name or other details about the experiment
An example of the leading comments:
: V1.0
: V4.0
: 10/06/2004
: aisaeed
: 10579
: IASCAG1
: 32448
:32448
: TIGR Spotfinder 2.2.3
: gpc30025a_532_nm.tif, gpc30025a_635_nm.tif
: Tumor type comparison
# This is the 4th experiment in a series of 20 to identify tissue-specific genes.
The header row consists of the field names for each subsequent row in this file (with the exception of comment lines). A minimum of seven columns must be present, and these must use a set of specifically named headers. Any number of additional columns may be included. The seven required column headers are:
:Unique identifier for this spot
: Intensity value in channel A
: Intensity value in channel B
: Row (slide row)
: Column (slide column)
: Meta-row (block row)
: Meta-column (block column)
As of version 4.0 of this file format the IA and IB columns can be substituted with MedA and MedB. The new requirement is that at least one integrated intensity (IA, IB, etc.) one median (MedA, MedB, etc.) value be reported for each channel in the microarray. For example, a two channel microarray .mev file would require either IA and IB MedA and MedB.
: Median intensity in channel A
: Median intensity in channel B
.mev files may use one of the following formats for the header row, depending on the origin of the mev file. The non-required columns (i.e. anything after the 7th column) may be rearranged and their names are subject to change at this time.
1. Database created mev file:
UID \t IA \t IB \t R \t C \t MR \t MC \t SR \t SC \t FlagA \t FlagB \t SAA \t SAB \t SFA \t SFB \t QCS \t QCA \t QCB \t BkgA \t BkgB
:Unique identifier for this spot
: Intensity value in channel A
: Intensity value in channel B
: Row (slide row)
: Column (slide column)
: Meta-row (block row)
: Meta-column (block column)
:Sub-row
: Sub-column
: TIGR Spotfinder flag value in channel A
: TIGR Spotfinder flag value in channel B
: Actual spot area (in pixels) in channel A
: Actual spot area (in pixels) in channel B
: Saturation factor in channel A
: Saturation factor in channel B
: Cumulative quality control score
: Quality control score in channel A
: Quality control score in channel B
: Background value in channel A
: Background value in channel B
2. Spotfinder created mev file:
UID \t IA \t IB \t R \t C \t MR \t MC \t SR \t SC \t FlagA \t FlagB \t SAA \t SAB \t SFA \t SFB \t QCS \t QCA \t QCB \t BkgA \t BkgB \t SDA \t SDB \t SDBkgA \t SDBkgB \t MedA \t MedB \t AID
:Unique identifier for this spot
: Intensity value in channel A
: Intensity value in channel B
: Row (slide row)
: Column (slide column)
: Meta-row (block row)
: Meta-column (block column)
:Sub-row
: Sub-column
: TIGR Spotfinder flag value in channel A
: TIGR Spotfinder flag value in channel B
: Actual spot area (in pixels) in channel A
: Actual spot area (in pixels) in channel B
: Saturation factor in channel A
: Saturation factor in channel B
: Cumulative quality control score
: Quality control score in channel A
: Quality control score in channel B
: Background value in channel A
: Background value in channel B
: Standard deviation for spot pixels in channel A
: Standard deviation for spot pixels in channel B
: Saturation factor in channel B
: Standard deviation of the background value in channel A
: Standard deviation of the background value in channel B
: Median intensity value in channel A
: Median intensity value in channel B
: Alternative ID
The first seven fields (UID, IA, IB, R, C, MR and MC) are required as specified above.
This flexible format allows users to track slide-specific data of interest, such as background, spot size and alternate intensities without requiring them of all users or adopting a limited ‘vocabulary’ of field names. This header row serves to identify the required and additional data columns. UID must be the left-most column in the mev file. Other columns do not need to be present in a fixed order.
For mev files generated at TIGR, the UIDs may be of the form: . For any given microarray database, the id field in the spot table will be unique. The combination of database and spot_id will therefore uniquely identify any spot on any array created at TIGR. It is important to note that this is not enough information to distinguish between spots in the same location on two slides of the same , as this would typically require an . Since annotation data is based on , it is not necessary to make this distinction, as all slides of a given type will use the same annotation file.
The AID column will usually contain an incremental sequence of numbers starting at 1. These can be used to return the file to the original sorted order and can function as a unique row identifier if necessary.
Applications that generate files of expression data (commonly in tav format) by retrieving records from the database access the spot table. , and are all capable of generating UIDs of the form described above in addition to the typical coordinate and intensity data.
mev files are required to end with the extension ‘.mev’. At this time there are no further naming conventions for mev files.
An annotation file is a tab-delimited text file containing annotation data for a specific . mev files can be associated with an annotation file only if both types of files are based on the same . The keys to this association are the unique ids in both files. Rows of mev and annotation files can be associated with each other if the unique ids are identical. A single header row is required to precede the annotation data in order to identify the columns below. Each remaining row of the file stores annotation data for a particular spot/feature on the array.
Annotation files may contain any number of non-computational comment lines. These lines, starting with ‘#’, will be treated identically to comment lines in mev files, and should precede the header row.
Annotation files created at TIGR will use UIDs that match the format used in the mev files, most likely . The structure of each annotation file is detailed below.
The header row consists of headers that identify each column of data. Each subsequent row of the file stores data for a particular spot/feature on the array. The annotation files created at TIGR will typically contain at least one comment at the top of the file with the following information:
: Version number based on revisions of annotation data
: The version of the .mev file format document
: Date of file creation or update
: Owner or the person responsible for creating the file
: Software tool used to create the document
: Version of the Gene Indices (or db?) that produced this
: Date of file creation or update
: type from the slide_type table that this array is based on
: Number of rows of annotation (eg. non-header) data
: Common name or other details about the experiment
An example of the leading comments:
: V3.0
: V4.0
: 04/20/2004
: jwhite
: 3.0
: IASCAG1
: 32448
: Database script
: Standard annotation file
The header row consists of the field names for each subsequent row in this file. Only the UID field is required. It must be the first field present and it must be named ‘UID’. Any number of additional fields may be included. Annotation files created at TIGR will always contain the following columns:
:Unique identifier for this spot
: Row (slide row)
: Column (slide column)
The remaining fields may vary, and a standard set has yet to be determined. Such a list will be published on a future date. R and C have been included to allow for manual alignment of the mev and corresponding annotation files in the event that the mev files were not generated in a traditional manner (ie. using Madam, etc.).
Some varieties of annotation files follow. The format may vary depending on the purpose of the file:
UID \t R \t C \t FeatN \t GBNum \t TCNum \t ComN \t …
UID \t R \t C \t GeneN \t Rxn \t PathwayN \t …
UID \t R \t C \t FeatN \t End5 \t End3 \t ChrNum \t …
Of course, it would be possible to combine the fields of these files, or add fields that have not been mentioned here. The goal is to keep the annotation flexible and the processing seamless.
There are not any naming conventions for annotation files at this time. If such a standard is introduced in the future, it will be detailed here.
A Bioconductor (MAS5) expression file is a tab-delimited text file that contains Affymatric Gene chip ID and several columns of expression datum. The header line contains all CEL file names you use in the Bioconductor calculation.
Sample_A.CEL Sample_B.CEL Sample_C.CEL 1053_at 435.013780957768 488.838904739281 435.013780957768 117_at 44.1563783495161 88.9787051028434 44.1563783495161 121_at 1222.87243433892 698.900718145166 1222.87243433892 1255_g_at 58.672587649119 47.0203292843508 58.672587649119 1294_at 336.502704708535 335.169154361150 336.502704708535 1316_at 163.254451578192 92.4927417614044 163.254451578192 1320_at 114.309125038560 105.223093019586 114.309125038560
Users can use following scripts to generate above files by using Bioconductor.
library(affy) data <-ReadAffy() mas5data<-mas5(data) write.exprs(mas5data,file="affy_mas5.txt") mas5call<-mas5calls(data) write.exprs(mas5call,file="affy_call.txt")
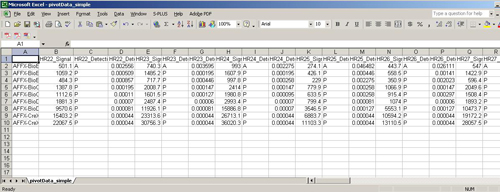
An Affymatrix GCOS file is a tab-delimited text file that contains Affymatric Gene chip ID and several experiment datum. In each experiment data it contains one column of intensity, one column of detection call and one column of p-value. The first line is head line.
GEO Simple Omnibus Format in Text (SOFT) file format is a kind of flexible tab delimited file format. Users can check the file format in details at this website.
A template for a single channel Sample file:
^SAMPLE=[required] !Sample_title = [required] !Sample_source_name = [required] !Sample_organism = [required] !Sample_characteristics = [required] !Sample_biomaterial_provider = [optional] !Sample_treatment_protocol = [optional] !Sample_growth_protocol = [optional] !Sample_molecule = [required][VALUE=total RNA, genomic DNA, polyA RNA, cytoplasmic RNA, nuclear RNA, protein, other] !Sample_extract_protocol = [optional] !Sample_label = [required] !Sample_label_protocol = [optional] !Sample_hyb_protocol = [optional] !Sample_scan_protocol = [optional] !Sample_description = [required] !Sample_data_processing = [required] !Sample_platform_id = [required; provide accession of existing GEO Platform for array used, e.g. "GPL96"] #ID_REF = [required; this column should correspond to the ID column of the reference platform] #VALUE = [required; typically supplied as normalized signal intensities] #HEADER_3 = [optional] #HEADER_4 = [optional] #HEADER_N = [optional; any number of user-defined columns can be included, and it is recommended that data tables be as comprehensive as possible excepting annotations that are provided on the platform entry] !Sample_table_begin ID_REF VALUE HEADER_3 HEADER_4 HEADER_N ...insert data table here; columns may appear in any order after the ID_REF column... !Sample_table_end
A template for platform file:
^PLATFORM=[required] !Platform_title = [required] !Platform_technology = [required][VALUE=spotted DNA/cDNA, in situ oligonucleotide, spotted oligonucleotide, antibody, tissue, MS, MPSS] !Platform_distribution = [required][VALUE=non-commercial, commercial, custom-commercial] !Platform_organism = [required] !Platform_manufacturer = [required] !Platform_manufacture_protocol = [required] !Platform_catalog_number = [optional] !Platform_support = [optional] !Platform_coating = [optional] !Platform_description = [optional] !Platform_web_link = [optional] !Platform_contributor = [optional; 1 per author; use 'firstname,lastname' or 'firstname,middleinitial,lastname'] !Platform_pubmed_id = [optional] #ID = [required; a unique id should be provided for each 'spot' on the array] #HEADER_2 = [required; all elements of the array should be identified using one or more columns in addition to the ID column] #HEADER_3 = [optional] #HEADER_N = [optional; provide as many headers as needed to fully describe the elements of the array] !Platform_table_begin ID HEADER_2 HEADER_3 HEADER_N ...insert data table here; columns may appear in any order after the ID column... !Platform_table_end
GEO Simple Omnibus Format in Text (SOFT) file format is a kind of flexible tab delimited file format for two channel data. Users can check the file format in details at this website.
A template for two channel file:
^SAMPLE=[required] !Sample_title = [required] !Sample_source_name_ch1 = [required] !Sample_organism_ch1 = [required] !Sample_characteristics_ch1 = [required] !Sample_biomaterial_provider_ch1 = [optional] !Sample_treatment_protocol_ch1 = [optional] !Sample_growth_protocol_ch1 = [optional] !Sample_molecule_ch1 = [required][VALUE=total RNA, polyA RNA, cytoplasmic RNA, nuclear RNA, genomic DNA, protein, other] !Sample_extract_protocol_ch1 = [optional] !Sample_label_ch1 = [required] !Sample_label_protocol_ch1 = [optional] !Sample_source_name_ch2 = [required] !Sample_organism_ch2 = [required] !Sample_characteristics_ch2 = [required] !Sample_biomaterial_provider_ch2 = [optional] !Sample_treatment_protocol_ch2 = [optional] !Sample_growth_protocol_ch2 = [optional] !Sample_molecule_ch2 = [required][VALUE=total RNA, polyA RNA, cytoplasmic RNA, nuclear RNA, genomic DNA, protein, other] !Sample_extract_protocol_ch2 = [optional] !Sample_label_ch2 = [required] !Sample_label_protocol_ch2 = [optional] !Sample_hyb_protocol = [optional] !Sample_scan_protocol = [optional] !Sample_description = [required] !Sample_data_processing = [required] !Sample_platform_id = [required; provide accession of existing GEO Platform for array used, e.g. "GPL1001"] #ID_REF = [required; this column should correspond to the ID column of the reference platform] #VALUE = [required; normalized log2 ratios are typically provided in this column] #HEADER_3 = [optional] #HEADER_4 = [optional] #HEADER_N = [optional; any number of user-defined columns can be included, and it is recommended that data tables be as comprehensive as possible excepting annotations that are provided on the platform entry] !Sample_table_begin ID_REF VALUE HEADER_3 HEADER_4 HEADER_N ...insert data table here; columns may appear in any order after the ID_REF column... !Sample_table_end
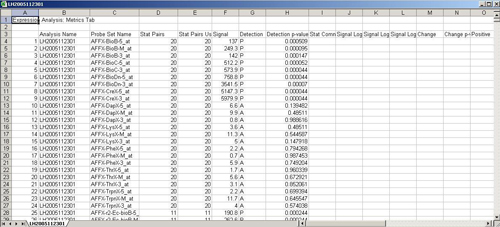
Each dChip data output file contains one experiment data. It’s a tab delimited file which records a lot of information for the experiment. Right now only chip ID, intensity, detection and detection p-value are read into Mev for the analysis.
Initialization dialogs for all modules save their information in a format that is logical, readable by other modules, and readable by humans.
Each module saves the header in the following way, with the abbreviated letters representing the module replacing *Module*.
# Assignment File
# User: user Save Date: Mon Jan 01 01:01:01 EST 2009
#
Module: *ModuleName*
MeV then stores a key which is essential for cross-compatibility between modules, as well as preserving label names for those modules that use labels.
Many modules contain a “not included” option for samples. This is not added to the key, and any assignment not associated with the key will be determined to be “not included”. This is important for cross-compatibility.
Group 1 Label: Alpha
Group 2 Label: Bravo
Group 3 Label: Charlie
The following lines are then written to the file for the purposes of human readability only.
#
Index Name Group Assignment
Each module will then write the group assignments for each sample, separated by a new line. For each assignment, at least 3 markers will be assigned, separated by tabs.
1.) The sample’s index, which will be assigned based on its location within the currently loaded data.
2.) The sample’s currently selected annotation, usually the default slide name.
3.) The sample’s group assignment that will match a String in the key above or a module defined “excluded” key.
Note: In some cases (BETR, NonPar, GSEA and TFA), additional information is stored for additional factors, conditions, etc. This information is added to the right of each key, and is ignored by other modules.
1 Sample A Alpha
2 Sample B Bravo
3 Sample C Charlie
4 Sample D Excluded
5 Sample E Alpha
6 Sample F Excluded
7 Sample G Alpha
8 Sample H Bravo
9 Sample I Charlie
The BETR module adds an additional parameter, “Conditions \t *int*”, which is necessary to tell the BETR reader whether or not to read condition assignments in addition to time assignments.
The reader does distinguish between files saved with “one-class” and “between subjects”. Though compatible, the user will be notified that the saved file was not saved for the current method. For “one-class” compatibility, only samples with a key in the first class are added to the “included” checkbox list. All others are excluded. “Paired” does not use this saving system.
Two-class paired:The loading file format is a tab-delimited file. Samples start from 0. Users define one pair in one line. Following is a sample file. The first sample and second sample are in one pair. The third sample and fourth sample are in one pair and so on.
0 1
2 3
4 5
6 7
The reader does distinguish between files saved with “one-class”, “multi-class” and “two-class unpaired”. Though compatible, the user will be notified that the saved file was not saved for the current method. For “one-class” compatibility, only samples with a key in the first class are added to the “included” checkbox list. All others are excluded. “Paired” and “Censored Survival” does not use this saving system.
Two-class paired:The loading file format is a tab-delimited file. Samples start from 0. Users define one pair in one line. Following is a sample file. The first sample and second sample are in one pair. The third sample and fourth sample are in one pair and so on.
0 1
2 3
4 5
6 7
Censored survival: The loading file format is a tab-delimited file. Users define one sample in one line. Following is a sample file. The first column is 1(selected) or 0(unselected). The second column is time and third column is 1(censored) or 0(dead).
1 0.0 0
1 0.0 1
1 0.0 1
1 0.0 1
Like BETR, TFA assignment files contain an additional column for keys belonging to the second factor. When loading other assignment files that do not contain 2 factors, only the first factor is assigned and the second remains unchanged.
In addition to loading group assignments, the saved group labels replace the currently loaded group labels. If the group label number does not match the current group label number, the loading process is stopped.
Like BETR and TFA, GSEA contains additional columns for additional factors. If the assignment file being loaded does not contain the additional columns needed to fill the additional factor assignments, GSEA will load only the first factor(s).
A “load file” feature has been added to the dialog box and is accessible from the “File” drop-down menu. All “excluded” samples from outside saved files are added to the “Neutral” group.
If a file is loaded with a different number of groups, MeV is able (at the user’s request) to replace the group label names with the saved names and add or subtract groups to match the saved file’s information.