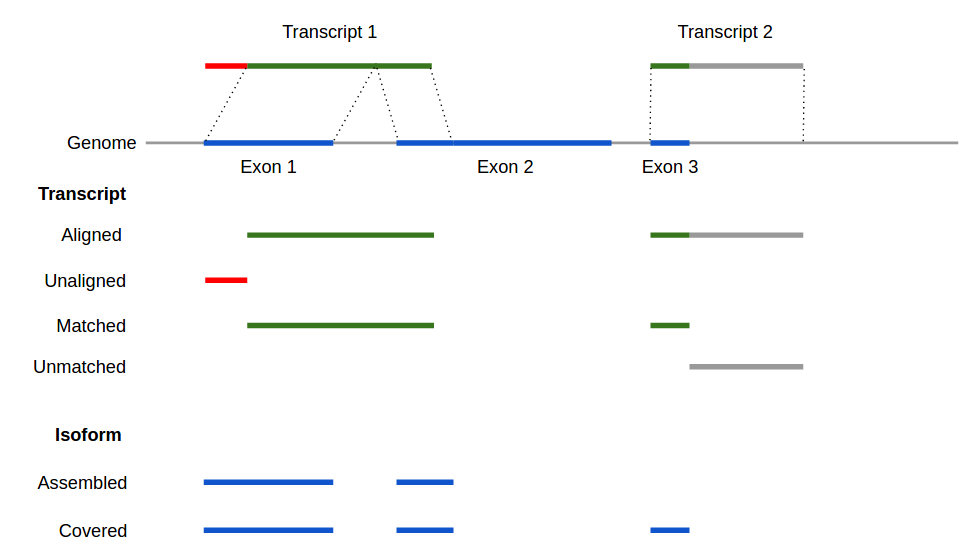
4.1 Reports
The following text files with reports are contained in comparison_output directory and include results for all input assemblies. In addition, these reports are contained in <assembly_label>_output directories for each assembly separately.
database_metrics.txt
Gene database metrics.
- Genes / Protein coding genes – number of genes / protein coding genes
- Isoforms / Protein coding isoforms – number of isoforms / protein coding isoforms
- Exons / Introns – total number of exons / introns
- Total / Average length of all isoforms, bp
- Average exon length, bp
- Average intron length, bp
- Average / Maximum number of exons per isoform
Coverage by reads. The following metrics are calculated only when --left_reads, --right_reads, --single_reads or --sam options are used (see options for details).
- Database coverage – the total number of bases covered by reads (in all isoforms) divided by the total length of all isoforms.
- x%-covered genes / isoforms / exons – number of genes / isoforms / exons from the database that have at least x% of bases covered by all reads, where x is specified with
--lower_threshold / --upper_thresholdoptions (50% / 95% by default).
basic_mertics.txt
Basic transcripts metrics are calculated without reference genome and gene database.
- Transcripts – total number of assembled transcripts.
- Transcripts > 500 bp
- Transcripts > 1000 bp
- Average length of assembled transcripts
- Longest transcript
- Total length
- Transcript N50 – a maximal number N, such that the total length of all transcripts longer than N bp is at least 50% of the total length of all transcripts.
alignment_metrics.txt
Alignment metrics are calculated with reference genome but without using gene database. To calculate the following metrics rnaQUAST filters all short partial alignments (see --min_alignment option) and attempts to select the best hits for each transcript.
- Transcripts – total number of assembled transcripts.
- Aligned – the number of transcripts having at least 1 significant alignment.
- Uniquely aligned – the number of transcripts having a single significant alignment.
- Multiply aligned – the number of transcripts having 2 or more significant alignments. Multiply aligned transcripts are stored in
<assembly_label>.paralogs.fastafile. - Misassembly candidates reported by GMAP (or BLAT) – transcripts that have discordant best-scored alignment (partial alignments that are either mapped to different strands / different chromosomes / in reverse order / too far away).
- Unaligned – the number of transcripts without any significant alignments. Unaligned transcripts are stored in
<assembly_label>.unaligned.fastafile.
Number of assembled transcripts = Unaligned + Aligned = Unaligned + (Uniquely aligned + Multiply aligned + Misassembly candidates reported by GMAP (or BLAT)).
Alignment metrics for non-misassembled transcripts
- Average aligned fraction. Aligned fraction for a single transcript is defined as total number of aligned bases in the transcript divided by the total transcript length.
- Average alignment length. Aligned length for a single transcript is defined as total number of aligned bases in the transcript.
- Average blocks per alignment. A block is defined as a continuous alignment fragment without indels.
- Average block length (see above).
- Average mismatches per transcript – average number of single nucleotide differences with reference genome per transcript.
- NA50 – N50 for alignments.
- Transcripts – total number of assembled transcripts.
- Misassembly candidates reported by GMAP (or BLAT) – transcripts that have discordant best-scored alignment (partial alignments that are either mapped to different strands / different chromosomes / in reverse order / too far away).
- Misassembly candidates reported by BLASTN – transcripts are aligned to the isoform sequences extracted from the genome using gene database with BLASTN and then transcripts that have partial alignments to multiple isoforms are selected.
- Misassemblies – misassembly candidates confirmed by both methods described above. Using both methods simultaneously allows to avoid considering misalignments that can be caused, for example, by paralogous genes or genomic repeats. Misassembled transcripts are stored in
<assembly_label>.misassembled.fastafile.
sensitivity.txt
Assembly completeness (sensitivity). For the following metrics (calculated with reference genome and gene database) rnaQUAST attempts to select best-matching database isoforms for every transcript. Note that a single transcript can contribute to multiple isoforms in the case of, for example, paralogous genes or genomic repeats. At the same time, an isoform can be covered by multiple transcripts in the case of fragmented assembly or duplicated transcripts in the assembly.
- Database coverage – the total number of bases covered by transcripts (in all isoforms) divided by the total length of all isoforms.
- Duplication ratio – total number of aligned bases in assembled transcripts divided by the total number of isoform covered bases. This metric does not count neither paralogous genes nor shared exons, only real overlaps of the assembled sequences that are mapped to the same isoform.
- Average number of transcripts mapped to one isoform.
- x%-assembled genes / isoforms / exons – number of genes / isoforms / exons from the database that have at least x% captured by a single assembled transcript, where x is specified with
--lower_threshold / --upper_thresholdoptions (50% / 95% by default). 95%-assembled isoforms are stored in<assembly_label>.95%assembled.fastafile. - x%-covered genes / isoforms – number of genes / isoforms from the database that have at least x% of bases covered by all alignments, where x is specified with
--lower_threshold / --upper_thresholdoptions (50% / 95% by default). - Mean isoform assembly – assembled fraction of a single isoform is calculated as the largest number of its bases captured by a single assembled transcript divided by its length; average value is computed for isoforms with > 0 bases covered.
- Mean isoform coverage – coverage of a single isoform is calculated as the number of its bases covered by all assembled transcripts divided by its length; average value is computed for isoforms with > 0 bases covered.
- Mean exon coverage – coverage of a single exon is calculated as the number of its bases covered by all assembled transcripts divided by its length; average value is computed for exons with > 0 bases covered.
- Average percentage of isoform x%-covered exons, where x is specified with
--lower_threshold / --upper_thresholdoptions (50% / 95% by default). For each isoform rnaQUAST calculates the number of x%-covered exons divided by the total number of exons. Afterwards it computes average value for all covered isoforms.
BUSCO metrics. The following metrics are calculated only when --busco and --clade options are used (see options for details).
- Complete – percentage of completely recovered genes.
- Partial – percentage of partially recovered genes.
GeneMarkS-T metrics. The following metrics are calculated when reference and gene database are not provided or --gene_mark option is used (see options for details).
- Genes – number of predicted genes in transcripts.
specificity.txt
Assembly specificity. To compute the following metrics we use only transcripts that have at least one significant alignment and are not misassembled.
- Unannotated – total number of transcripts that do not cover any isoform from the database. Unannotated transcripts are stored in
<assembly_label>.unannotated.fastafile. - x%-matched – total number of transcripts that have at least x% covering an isoform from the database, where x is specified with
--lower_threshold / --upper_thresholdoptions (50% / 95% by default). - Mean fraction of transcript matched – matched fraction of a single transcript is calculated as the number of its bases covering an isoform divided by the transcript length; average value is computed for transcripts with > 0 bases matched.
- Mean fraction of block matched – matched fraction of a single block is calculated as the number of its bases covering an isoform divided by the block length; average value is computed for blocks with > 0 bases matched.
- x%-matched blocks – percentage of blocks that have at least x% covering an isoform from the database, where x is specified with
--lower_threshold / --upper_thresholdoptions (50% / 95% by default). - Matched length – total number of transcript bases covering isoforms from the database.
- Unmatched length – total alignment length - Matched length.
relative_database_coverage.txt
Relative database coverage metrics are calculated only when raw reads (or read alignments) are provided. rnaQUAST uses read alignments to estimate the upper bound of the database coverage and the number of x-covered genes / isoforms / exons (see read coverage) and computes the following metrics:
- Relative database coverage – ratio between transcripts database coverage and reads database coverage.
- Relative x%-assembled genes / isoforms / exons – ratio between transcripts x%-assembled and reads x%-covered genes / isoforms / exons.
- Relative x%-covered genes / isoforms / exons – ratio between transcripts x%-covered and reads x%-covered genes / isoforms / exons.
4.3 Detailed output
These files are contained in <assembly_label>_output directories for each assembly separately.
<assembly_label>.unaligned.fasta– transcripts without any significant alignments.<assembly_label>.paralogs.fasta– transcripts having 2 or more significant alignments.<assembly_label>.misassembled.fasta– misassembly candidates detected by methods described above. Seemisassemblies.txtdescription for details.<assembly_label>.correct.fasta– transcripts with exactly 1 significant alignment that do not contain misassemblies.<assembly_label>.x%assembled.list– IDs of the isoforms from the database that have at least x% captured by a single assembled transcript, where x is specified by the user with an option--upper_threshold(95% by default).<assembly_label>.unannotated.fasta– transcripts that do not cover any isoform from the database.
4.3 Plots
The following plots are similarly contained in both comparison_output directory and <assembly_label>_output directories. Please note, that most of the plots represent cumulative distributions and some plots are given in logarithmic scale.
Basic
transcript_length.png– assembled transcripts length distribution (+ database isoforms length distribution).block_length.png– alignment blocks length distribution (+ database exons length distribution).x-aligned.png– transcript aligned fraction distribution.blocks_per_alignment.png– distribution of number of blocks per alignment (+ distribution of number of database exons per isoform).alignment_multiplicity.png– distribution for the number of significant alignment for each multiply-aligned transcript.mismatch_rate.png– substitution errors per alignment distribution.Nx.png– Nx plot for transcripts. Nx is a maximal number N, such that the total length of all transcripts longer than N bp is at least x% of the total length of all transcripts.NAx.png– Nx plot for alignments.
Sensitivity
x-assembled.png– a histogram in which each bar represents the number of isoforms from the database that have at least x% captured by a single assembled transcript.x-covered.png– a histogram in which each bar represents the number of isoforms from the database that have at least x% of bases covered by all alignments.x-assembled_exons.png– a histogram in which each bar represents the number of exons from the database that have at least x% captured by a single assembled transcript.x-covered_exons.png– a histogram in which each bar represents the number of exons from the database that have at least x% of bases covered by all alignments.alignments_per_isoform.png– plot showing number of transcript alignments per isoform
Specificity
x-matched.png– a histogram in which each bar represents the number of transcripts that have at least x% matched to an isoform from the database.x-matched_blocks.png– a histogram in which each bar represents the number of all blocks from all transcript alignments that have at least x% matched to an isoform from the database.
5 Citation
Bushmanova, E., Antipov, D., Lapidus, A., Suvorov, V. and Prjibelski, A.D., 2016. rnaQUAST: a quality assessment tool for de novo transcriptome assemblies. Bioinformatics, btw218.
6 Feedback and bug reports
Your comments, bug reports, and suggestions are very welcomed. They will help us to further improve rnaQUAST. If you have any troubles running rnaQUAST, please send uslogs/rnaQUAST.log from the output directory.
Address for communications: rnaquast_support@ablab.spbau.ru.