Bill Pearson's FASTA Web site: [http://www.people.Virginia.EDU/~wrp/pearson.html]
Retrieve PIR entry A34313 to a file called A34313.pir. To read this sequence into GDE, choose ' File --> Import Foreign Format' and type in A34313.pir.
(Note: the File --> Open menu can only read 3 formats: GenBank, GDE flatfile, and GDE). All other formats, except for FSAP Free Format, must be read in using File --> Import Foreign Format).
Retrieve GenBank entry J02593 to a file called SRAAFP.gen. To read this sequence into GDE, choose 'File --> Open', and select SRAAFP.gen.
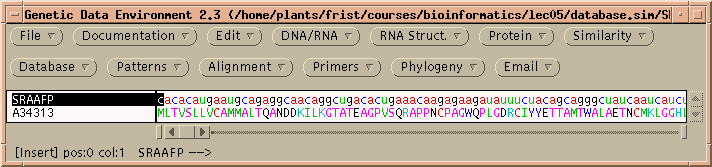
These two files are respectively, the DNA and protein sequences for an antifreeze protein from the sea raven, Hemitripterus americanus. Both sequences should now be in your GDE window.

The Database menu contains four programs from the FASTA package:

First, choose a database. All GenBank divisions are listed, along
with
a choice to search ALL divisions, or a User-created file in
Pearson/FASTA
format. Most of the time, it is best to search the taxonomic group from
which your query sequence is derived. This is especially true for
DNA/DNA
searches, since only closely-related DNA sequences are likely to give
statistically-significant
matches. If you do need to search all of GenBank, this can take
more
than an hour, depending on the length of the sequence and the K-tuple
value
(each K-tuple speeds up the search by a factor of 4, but decreases
sensitivity).
| Hint: Send
output from long-running
searches to a file.
GDE runs all database searches in the background, ie. as independent jobs. If you quit GDE or even logout, the job will run to completion. For long-running searches, it is usually best to send output directly to a file. Simply click on 'Output file', and type an Output file name. In the example above, output will be directed to two files: SRAAFP.gbvrt.fasta, which contains the FASTA report, and SRAAFP.gbvrt.fasta.nam, which contain the names of the hits. This namefile could be used as input to FETCH to retrieve the hits. Notice that GDE automatically appends the .nam file extension. |
| Hint: For very
closely-related
sequences, speed up the search with K-tuple = 5 or 6.
In some cases, you are specifically looking for sequences that share very high similarity with your query. Suppose you had a cDNA and wanted to find the corresponding genomic clone. In that case, the similarity is likely to be greater than 90%. A K-tuple value of 5 or 6 will give a very fast search at low sensitivity, which is all you need. |

The menu shows that you can search either the PIR database, GenPept, which is translated protein coding sequences from GenBank, or a User-created file containing amino acid sequences in Pearson/FASTA format.

The menu shows a variety of protein scoring matrices. The Blosum matrices were constructed using distantly-related sequences. If you need a highly sensitive search, use a low-numbered Blosum matrix. The PAM matrices were constructed from a set of closely-related sequences. For a high-sensitivity search, use PAM250, or PAM250 Gonnet (based on a more recent dataset.). For closely-related sequences, use PAM120.
More information of scoring matrices can be found in
Hugh B. Nicholas Jr., David W. Deerfield II., and Alexander J.
Ropelewski, (1998) A Tutorial on Searching Sequence Databases and
Sequence
Scoring Methods Developed by the Biomedical Supercomputing
Initiative
of the Pittsburgh Supercomputing Center.[http://www.psc.edu/biomed/training/tutorials/sequence/db/].
The results of this protein search are found in A34313.pir.fasta
and
A34313.pir.fasta.nam.
Hint: Oligopeptides need logarithmic score weightingFor very short query sequences such as oligopeptides, unweighted scores would not exceed the minimum cutoff values needed to appear in the output. The FASTA programs allow you to specify a logarithmic weighting ratio that gives short query sequences higher matches. The score is weighted by the natural log of the query length divided by the natural log of the database size. |

TFASTA is the most sensitive method for searching DNA sequence databases. Each DNA sequence is translated on the fly in either 3 or 6 reading frames (ie. 1 or both strands) . ALL sequence is translated, whether coding or non-coding, intron or exon. TFASTA does a simple translation. TFASTX allows 3-base insertions (ie. 1 codon), while TFASTY allows 1 or 2 base insertions. TFASTY is therfore the most sensitive program, but it is also slower.TFASTY would be especially good at detecting similarities between a query protein and ESTs, since ESTs typically have more frameshifts than most sequences.
The results of this protein search are found in A34313.gbvrt.tfasta
and
A34313.gbvrt.tfasta.nam.
| Note on automatic translation: Programs that translate sequences on the fly (eg. TFASTA, TFASTX, TBLASTN, TBLASTX) have no knowledge whatsoever about gene structure (ie. exons, introns, 5' UTR, 3'UTR). All these programs do is to take every group of 3 nucleotides and assign a codon to it. Even stop codons are represented by an asterisk (*). Consequently, non-coding sequences and non-coding open reading frames are transated into meaningless amino acid sequences. |
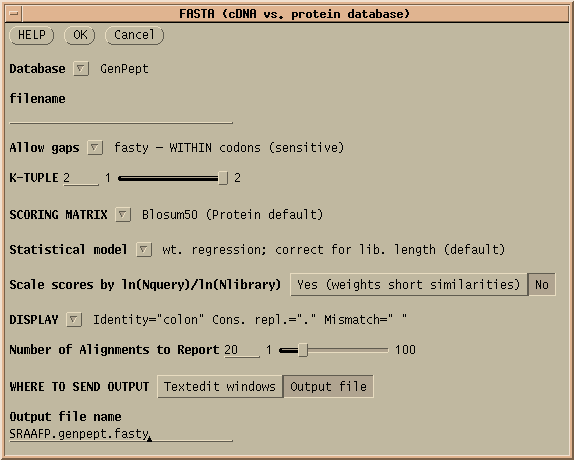
FASTX and FASTY are the converse of TFASTX and TFASTY, in that they translate a DNA query sequence, allowing either codon-sized gaps (TFASTX) or 1 or 2 base gaps (TFASTY). TFASTY is therefore the slower, but more sensitive. These programs are particularly well-suited for comparing a sequence that may have frameshifts (eg. an EST) with a protein database.
The results of this protein search are found in SRAAFP.genpept.fasty
,
SRAAFP.genpept.fasty.gb, and SRAAFP.genpept.fasty.gi. The .gb
and .gi extensions indicate that these files contain GenBank ACCESSION
numbers and NCBI GI numbers, respectively. Either list could be used to
retrieve the hits from GenBank.
