TUTORIAL:
Extracting features from text files
|
Sep. 16, 2022 |
TUTORIAL:
Extracting features from text files
|
Sep. 16, 2022 |
| artemis |
launch artemis |
| Go to the Feature window at the bottom of Artemis and right click on the mouse. To save these features in a file, right-click and choose Save List to File. Name the file Acidianus_hospitalis_fea.txt. |  |
| step |
command |
explanation |
1
|
grep CDS < Acidianus_hospitalis_fea.txt > out1.txt | grep reads lines from
Acidianus_hospitalis_fea.txt and prints only those lines
containing the string 'CDS' to out1.txt. |
2
|
cut -c 1-33 < out1.txt
> out2.txt |
Cut out columns 1 through 33 from out1.txt
and write them to out2.txt. |
3
|
tr -s ' ' < out2.txt > out3.txt |
tr reads lines from out1.txt and prints
those lines with all multiple blanks translated into
single blanks (the -s option) |
4
|
tr ' ' '\t' < out3.txt > out4.txt |
tr reads lines from out2.txt and prints to
out3.txt those lines with each blank translated into a tab
character, represented by \t in the command. Both \t
and the blank must be enclosed in quotes. |
5
|
sed -e 's%\t$%\tf%' < out4.txt > Acidianus_hospitalis_fea.tsv | The Stream Editor, sed, reads lines from
out3.txt and prints those lines, modified according
to the expression after the -e option. This expression
says substitute (s) each occurrence of tab followed by the
end of a line (\t$) with a tab followed by the letter f.
Output is written to Acidianus_hospitalis_fea.tsv. |
| Doing the task as a
single command - Remember that in Unix, pipes (|) tell
the shell to redirect the standard output from one command
the standard input for the next command. This eliminates the
need to save intermediate steps in temporary files. The
series of steps listed above could be precisely replicated
with the following command line: cat Acidianus_hospitalis_fea.txt | grep CDS | cut -c 1-33 | tr -s ' ' \ | tr ' ' '\t' | sed -e 's%\t$%\tf%' > Acidianus_hospitalis_fea.tsv Note: when a command is very long, you can break it up into two or more lines by inserting a left slash '\', which tells the shell to continue reading on the next line. |
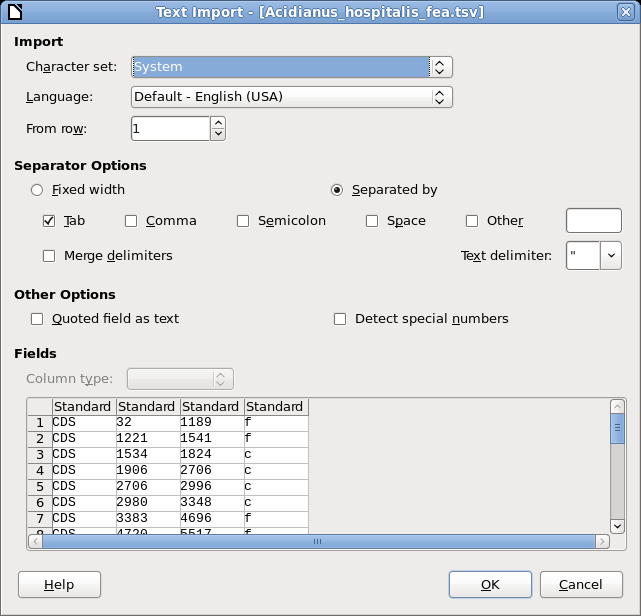
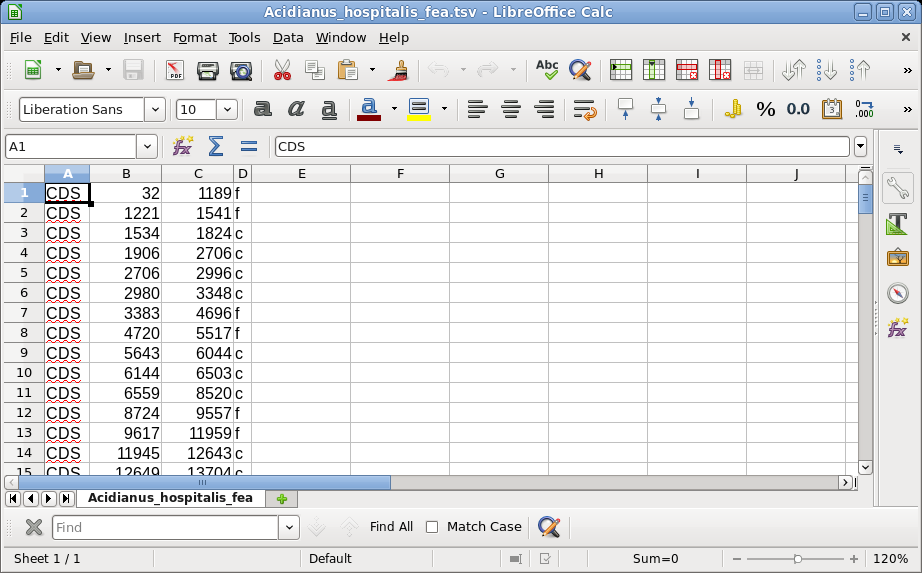
| Not all text files are
easily machine-parsable - Like many bioinformatics
programs, the text files in which Artemis saves feature
information were not explicitly designed to be
machine-parsable. For example, when Artemis saves features
from the Zea mays entry KR014666, an excerpt from the file
has the following lines: gap 1063651 1063750 gap 1071134 1071233 CDS 1091603 1093045 c similar to B73 protein GRMZM2G010987_T01 mRNA <1091603 >1093045 c CDS 1109880 1110602 c similar to B73 protein GRMZM5G835704_T01 mRNA <1109880 >1110602 c gap 1114991 1115090 gap 1124793 1124892 gap 1127331 1127430 gap 1131616 1131715 The fixed column format that Artemis uses did not take into account the GenBank convention that uses < and > to indicate that the precise start and stop sites of a feature are unknown, but are assumed to be respectively upstream and downstream of the listed coordinates. Since these characters shift columns 4 and 5 to the right, the cut command we used above would not correctly cut out the first 4 columns. A similar problem would occur if features were saved for sequences greater than 9,999,999 nucleotides in length. For a more general solution to parsing these files, more sophisticated pattern recognition methods, probably involving regular expressions, would have to be employed. |