Click on the filename, and click 'Open'.
{brassica:/home/plants/frist}cd
do next step
if $HOME/bioinf doesn't
exist
{brassica:/home/plants/frist}mkdir
tutorials
create directory for this
tutorial
{brassica:/home/plants/frist}mkdir
tutorials/sequence
copy GenBank files to new directory
{brassica:/home/plants/frist}cd
$birch/tutorials/bioLegato/sequence
{brassica:/home/plants/frist}cp
*.gen $HOME/tutorials/sequence
return to $HOME directory verify that
new files and directories are present
{brassica:/home/plants/frist}cd
{brassica:/home/plants/frist}ls
-l
drwx------
1
frist drr 512 Oct 31 10:11
tutorials/
{brassica:/home/plants/frist}cd
tutorial
{brassica:/home/plants/frist/tutorials}ls
-l
drwx------
3
frist drr 512 Oct 31 10:11
sequence/
{brassica:/home/plants/frist/tutorials}cd
sequence
{brassica:/home/plants/frist/tutorials/sequence}ls
-l
-rw-------
1 frist frist
127286 Oct 31 10:13 AC002329.gen
-rw------- 1 frist frist 5404
Oct
31 10:13 ARBLKSP.gen
-rw------- 1 frist frist 10739 Oct
31
10:13 PBI101TD.gen
-rw------- 1 frist frist 8278
Oct
31 10:13 pBSGUS.gen
-rw------- 1 frist frist 3674
Oct
31 10:13 PEACAB15.gen
Files with the .gen extension are in GenBank format. Since these are
ASCII
text files, you can view them in any text editor. Double clicking on a
file
in the file manager will bring up the file in the default text editor
for your bioLegato installation.
Example: Reading and printing PEACAB15.gen with NUMSEQ
Name: PEACAB15 Topology: LINEAR Length: 822 ntBy default, NUMSEQ will print out the entire sequence (from START to FINISH) as a single strand (STRANDS) in 7 groups (GPL) of 10 nucleotides (GROUP) per line. To change parameters, type the number of a parameter, and you will be prompted for a new value. When you're ready to view the sequence with the new parameters, type '0' at the prompt and '5' in the main menu to print the sequence to the screen.
________________________________________________________________________________
Parameter Description/Response Value
________________________________________________________________________________
1)START first nucleotide printed 1
2)FINISH last nucleotide printed 822
3)NUCCASE U:(A,G,C,T...), l:(a,g,c,t...) U
4)STARTNO number of starting nucleotide 1
5)GROUP number every GROUP nucleotides 10
6)GPL number of GROUPs printed per line 7
7)WHICH I: input strand O: opposite strand I
8)STRANDS 1: one strand, 2:both strands 1
9)KIND R:RNA D:DNA D
10)NUMBERS Number the sequence (Y or N) Y
11)NUCS Print nucleotide seq. (Y or N) Y
12)PEPTIDES Print amino acid seq. (Y or N) N
13)FRAMES 1 for this frame, 3 for 3 frames 1
14)FORM L:3 letter amino acid, S: 1 letter L
________________________________________________________________________________
Type number of parameter you wish to change (0 to continue)
To view both strands:
8) STRANDS: 2To translate in 3 reading frames:
12) PEPTIDES: yTo limit printing to only part of the seuqence eg. bases 200 - 400:
13) FRAMES: 3
5) GROUP: 15
6) GPL: 4NUMSEQ breaks up the sequence into groups of nucleotides, numbering each group. For translation, GROUP must be divisible by 3, because translation is done in discrete codons of 3 bases each. GPL is set to 4 so that the output line will fit on a typical 80-character line.
1) START: 200To view the opposite strand of the same region:
2) FINISH: 400
7) WHICH: o
1) START: 400
2) FINISH: 200This example illustrates that creating an opposite strand requires two steps. First, we have to specify the strand as 'o' (opposite) rather than 'i' (input strand). This causes the bases to be complemented. However, if all we do is complement the input strand, then the opposite strand would be printed 3' to 5', because we would be starting at 200 and ending at 400. Therefore, START must be set to 400, and FINISH to 200.
To illustrate the point, let's try running NUMSEQ from bioLegato.
Launch bioLegato from the command line.
{brassica:/home/plants/frist/tutorials/sequence}biolegato
| IMPORTANT NOTES:
1.While bioLegato is running, the terminal window can not be used for other commands. If you need to type commands, open another terminal window. 2. Although bioLegato can read files from any directory, it's best to launch bioLegato from the directory in which you plan to work. |
Read in PEACAB15.gen:
File --> Open
Click on the filename, and click 'Open'.

Hint: There are 2
steps
to running a program from bioLegato
|
To run numseq, click on PEACAB15 and choose DNA/RNA --> NUMSEQ.
The numseq menu appears, containing menu items for all parameters in the NUMSEQ Parameters menu.
HINTS ON bioLegato MENUS:
|

Nrmally, the temporary output file (eg. bioLegato74903554674472742396.tmp.out) will be deleted when you quit the Text Editor window. To save the file, choose File --> Save As and type in a name for the output. It's a good idea to include a .numseq file extension to indicate that this file is output from numseq.
Because the output is ASCII text, you can do lots of things with it,
including
importintg it into a word processor, pasting it into another window,
mailing
it, or even using it as input for other sequence programs. In the
latter
case, the output will probably need to be modified to conform to the
desired
input file format eg. Pearson/Fasta.

Circular DNA molecules require a bit of thought. Since printing is always done 5' --> 3', the direction (clockwise vs. counterclockwise) determines the strand, or vice versa. Consider the Bluescript cloning vector (GenBank X52331). Conceptually, one base must be arbitrarily labeled as 1. In the GenBank entry, 1 is the first base in the file, and 2958 is the last base in the file. In the physical plasmid, of course, base 2958 is adjacent to 1.
In NUMSEQ, the START, FINISH and WHICH parameters govern which parts of the sequence are displayed.
To view the top strand of the PvuI (CGAT^CG) fragment going clockwise from 2417 to 503:
1) START: 2417Since you're only considering 1 strand at a time, you want to start with 2417, which is the 5' end of the small PvuI fragment, on the original strand.
2) FINISH: 503
7) WHICH: Original
To print the same sequence on the other strand, we can't just change WHICH to 'Opposite".
1) START: 2417Try it and you'll see that what you get is the large PvuI fragment going from 2417 to 503, and that this fragment doesn't even terminate where PvuI would cut. It's best to visualize the fragment ends as illustrated below:
2) FINISH: 503
7) WHICH: Opposite
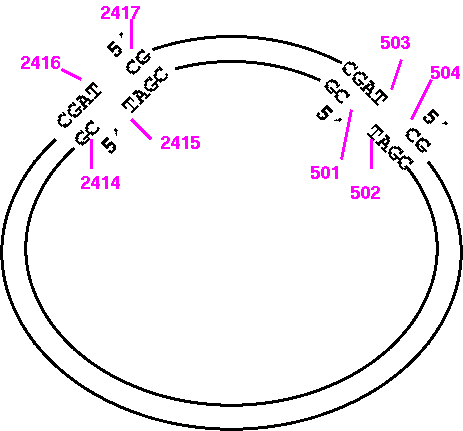
So the correct way to print the opposite strand of the small fragment would be:
1) START: 501Example: Simulated restriction digest of a pBluescriptKSm13+ at BamH1
2) FINISH: 2415
7) WHICH: Opposite
1) START: 690If we wanted the inverse complement (ie. counter clockwise), the NUMSEQ parameters would be
2) FINISH: 689
7) WHICH: Original
1) START: 693
2) FINISH: 694
7) WHICH: Opposite
EXAMPLE: Cloning beta-glucuronidase gene (GUS) from pBI101 to pBluescriptKSm13+.
The GUS gene in pBI101 can be conveniently excised using BamHI and
SacI (see map).
The goal is to make a datafile that correctly represents the
recombinant construct that results from cloning the BamHI/SacI fragment
containing the GUS gene into the BamHI/SacI-digested BlueScript
plasmid. It should look something like this:
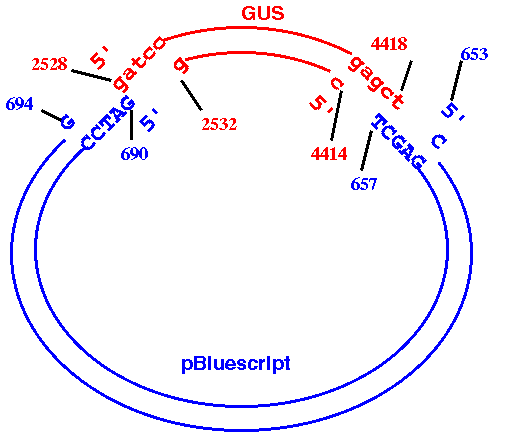
How to do it:
a. Read GenBank entries for pBI101 (PBI101TD.gen) and pBluescriptKSm13+ (ARBLKSP.gen) into bioLegato.b. Use DNA/RNA --> BACHREST to find the locations of the BamHI and SacI sites in PBI101TD. (PBI101TD BACHREST output). According to BACHREST, the 5' ends of the BamHI and SacI sites are at 2528 and 4419, respectively. Therefore, the 3' end of the fragment we want is at 4418, not 4419.
(See 'II. What the output means' in the BACHREST documentation file rest.asc for details on the output.)
c. PI101TD --> NUMSEQ
START: 2528d. Use DNA/RNA --> BACHREST to find the locations of the BamHI and SacI sites in ARBLKSP. (ARBLKSP BACHREST output). According to BACHREST, the 5' ends of the BamHI and SacI sites are at 690 and 658, respectively. Since the GUS fragment terminates at a SacI site, the SacI site from the plasmid must come next, with the BamHI site at the other end. We need to generate the opposite strand of the plasmid, going from the 5' end of SacI to the 3' end of BamHI.
FINISH: 4418Save this output in pBSGUS.dna, and minimize the window to get it out of the way.
e. ARBLKSP --> NUMSEQ
START: 653f. Before going any farther, verify that the construct has been built correctly. One way to do this is to use NUMSEQ to generate double-stranded printouts of both original sequences, and then mark the positions of the restriction sites on these printouts. Print out pBSGUS.dna and compare the sequence at the cloning junctions to the sequences in the originals. Make sure that complete BamHI and SacI sites appear at these junctions.
FINISH: 694
WHICH: OppositeCopy this output to the end of pBSGUS.dna and save the fille.
g. Convert pBSGUS into a Pearson/Fasta file to be read by SEQUIN.
In bioLegato, use File --> Import Free Format, and type in 'pBSGUS.dna' Note that free-format files do not contain sequence names, so the filename is used as the sequence name, in bioLegato. Since we don't want the .seq extension to be part of the name, get rid of '.dna' in File --> GetInfo. The name should now be 'pBSGUS'.h. The last step in creation of a sequence file is annotation. This is critical, because it documents precisely what you have done. The ability to reproduce results is as important in computers as it is in the lab. GenBank format is the richest and most versatile sequence file format, and it is read by most sequence programs. SEQUIN automates the process of creating GenBank format files.Save as a Pearson/Fasta file by choosing File --> Export Foreign Format, and typing 'pBSGUS.wrp'.
The menus in SEQUIN walk you throught a step-by-step process of the minimal information needed for a GenBank entry. Without going into every step, the over all series of events is as follows:
1. Start SEQUIN by typing 'sequin' at the command line.
2. Choose "Start new submission"
3. Fill in information in the Submission, Contact, Author and Title (all required)
4. On the page entitled Organism and Sequences, click on "Import Nucleotide FASTA" to import your .wrp file
5. Click on "Specify Topology" and set the topology to 'Circular'.
6. For Organism, type in 'synthetic construct'.
7. This is the minimum information needed to create a GenBank entry that can be used as a model for a restriction digest. Once the minimal information has been entered, follow the 'Next Page' links until a window pops up with the GenBank entry in it. (For other purposes, you may wish to annotate locations of coding sequences and other features of interest. In a laboratory setting, if you were planning to submit the sequence to GenBank, the most critical things to annotate for a construct such as this are the precise sources of the component sequences, in the FEATURES TABLE, along with a simple explanation in words in the DEFINITION line.)
8. To export your sequence to a GenBank file, choose File --> Export GenBank. Save your sequence as pBSGUS.gen.A good introduction to SEQUIN, including screen shots, can be fund at http://www.ncbi.nlm.nih.gov/Sequin/.
i. Test your GenBank file by reading it into bioLegato, and running BACHREST. The BACHREST output should show that pBSGUS is circular, and the BamHI and SacI sites at 1 and 1892, respectively (pBSGUS.bachrest).