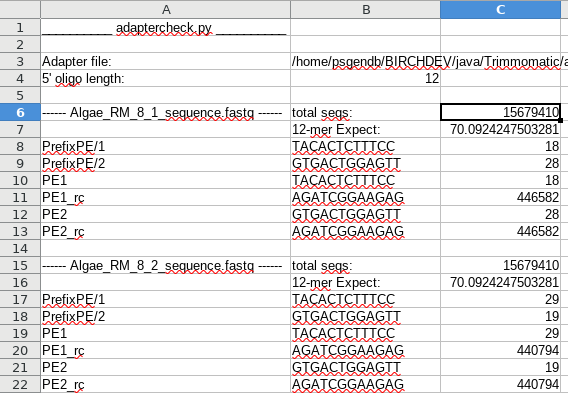
| An example of output is shown at right. For each fastq file, the total number of sequences is listed, along with the expected number of hits for a k-mer of a specified size. In this example, out of 15,679,410 sequences, we'd expect to see 70 hits of 12 nt in length. This is based on the assumption that the a hit will occur once every 4k nucleotides. For most adapters, the observed number of hits is between 18 and 29. In the first file, we see that both PE1_rc and PE2_rc were found in 446,582 reads. These almost certainly represent real read throughs. Dividing the number of hits by the total number of sequences, we estimate that about 2.8% of these reads are read-throughs, in which at least 12 bp of adapter are at the 3' end of the read. |
 |