TUTORIAL: SEARCHING LOCAL SEQUENCE DATABASES USING FASTA |
Apr. 20, 2020 |
TUTORIAL: SEARCHING LOCAL SEQUENCE DATABASES USING FASTA |
Apr. 20, 2020 |
This
tutorial assumes that local copies of the NCBI RefSeq RNA,
RefSeq protein, Non-redundant nudcleotide (nt) and
Non-redundant protein (nr) databases.
|
This tutorial will introduce the several
types of sequence database searches, using the antifreeze protein from the sea raven, Hemitripterus
americanus. The cDNA sequence for this gene is found in SRAAFP.gen.
Create a directory called 'db' and save
this sequence to the db directory.
To read this sequence into bldna, choose 'File --> Open', and select SRAAFP.gen.
|
Since this tutorial will demonstrate database searches
using both DNA and protein sequences as query sequences,
we need to generate the amino acid sequence. This is done
in two steps. First, we need to extract the protein coding
sequence from the cDNA. (Remember that mRNAs contain both
5' and 3' untranslated sequences. If you look at SRAAFP.gen,
you'll see that the CDS (protein coding sequence) runs
from positions 10 to 597. We will use the FEATURES program
to extract the CDS, and then translate the CDS using
RIBOSOME. |
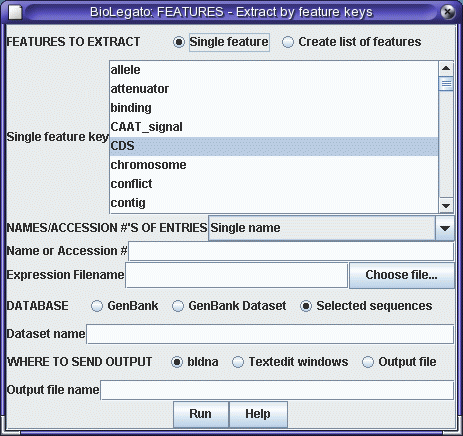 |
Click on 'Run' with the above settings. The CDS will appear in a
new bldna window.
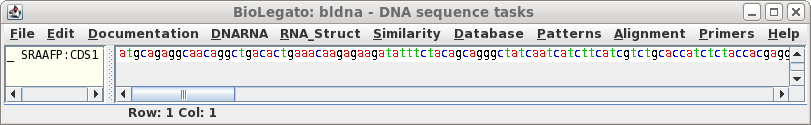
|
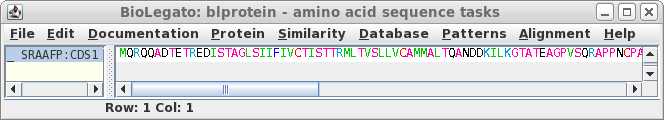
Note that the CDS begins with a start codon, indicating
that this cDNA contains the entire protein coding region. Next translate the CDS into protein. Select SRAAFP:CDS1, and choose DNARNA --> Ribosome. By default, Ribosome sends the output to a new blprotein window. |
 |


| First, choose a database. The quickest one to search is probably RefSeq RNA, which contains the transcripts and coding regions from most known genes encoding RNAs and proteins. Non-coding genomic regions, which make up the vast majority of eukaryotic genomes, are omitted from this dataset, which is consequently a lot smaller and faster to search. | 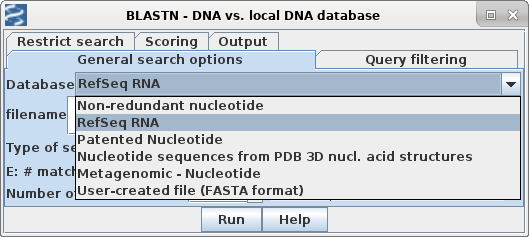 |
| How many threads should
I use? FASTA can break the database search into 2 or more threads which work simultaneously on different parts of the problem. The more threads, the faster the search will be completed. Most desktop computers and laptops will have at least 2 or 4 CPUs, while the example at right is taken from Biolegato running on a server with 64 CPUs. BioLegato will set the default number of threads to 1/4 of the number of CPUs available on your computer. If you increase the number of threads, always use at least some free CPUs for other tasks on the system. In a workshop setting where many students are running FASTA searches at the same time, it is best to limit each student to a few threads. Each search will take longer, but that is a trade-off to avoid an overall degradation of system performance. |
| Hint:
For more distantly-related sequences, use K-tuple =
4 or 5. The default K-tuple value of 6 ensures a faster search. However, some sensitivity is lost by using a larger K-tuple value. If you are concerned about finding sequences that are more distantly-related to your query, use K-tuple 4 or 5. |
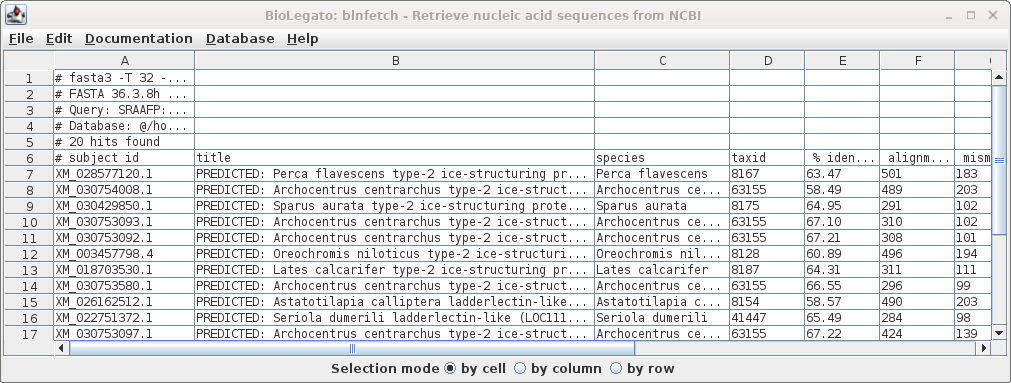
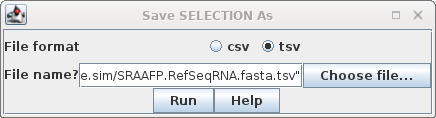
| Similarly, the list of accession numbers can
be saved from blnfetch by choosing File --> Save
Selection As SRAAFP.RefSeqRNA.fasta.tsv. Note that the file format was set to TSV, but the file extension is .acc. Since the file only contains a single column, the distinction between a TSV file and a text file is moot. |
 |
| A quick word on CSV, TSV and text files,
and BioLegato Most spreadsheet programs can read and write formats called CSV and TSV. CSV files are comma-separated value text files, in which each line in the file represents a row in a spreadsheet. The values for each column in a row are separated by commas. TSV files are TAB-separated value text files. In these files, values in each row are separated by TAB characters. TSV files are probably safer to use, because CSV files make it impossible to include commas within fields. BioLegato programs such as blnfetch, blpfetch and blncbi have a table canvas, that works like a spreadsheet. In these programs, the Open menu can only read text files with a .csv or .tsv file extension. To read files with other extensions eg. .acc, you need to use File --> Import Data from CSV file. Text files such as the one generated above will be read using this approach. |
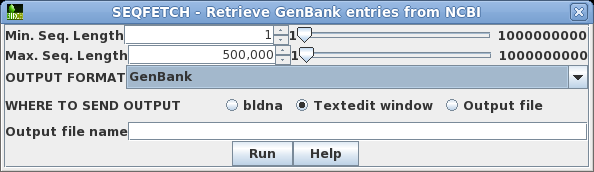
| Retrieving your hits If you decide to retrieve all the hits, this can be done from blnfetch. Choose Edit --> Select All, and then Database --> SeqFetch. By default, SEQFETCH is set to retrieve GenBank entries to a text editor. If you had a large number of entries, it would probably be best to sent output to a file. |
 |
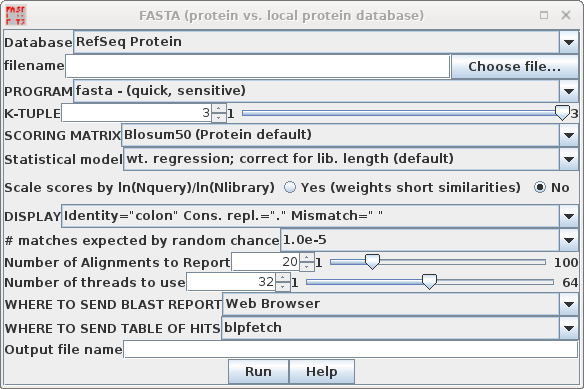
| In your blprotein window, select the amino
acid sequence SRAAFP:CDS1, and choose Database -->
FASTA (protein vs. protein database). Note that by default, accession numbers will be sent to blpfetch, a BioLegato interface for retrieving protein entries, while the FASTA report will go to a text editor.
|
 |
 For DNA searches a very simple
scoring scheme is used, eg. +1 is added to the score for a match,
and -1 is added to the score for a mismatch. The greater diversity
of amino acids lends itself to more complex scoring schemes. The
menu shows a variety of protein scoring matrices. The Blosum
matrices were constructed using distantly-related sequences. If
you need a highly sensitive search, use a low-numbered Blosum
matrix. The PAM matrices were constructed from a set of
closely-related sequences. For a high-sensitivity search, use
PAM250, or PAM250 Gonnet (based on a more recent dataset.). For
closely-related sequences, use PAM120.
For DNA searches a very simple
scoring scheme is used, eg. +1 is added to the score for a match,
and -1 is added to the score for a mismatch. The greater diversity
of amino acids lends itself to more complex scoring schemes. The
menu shows a variety of protein scoring matrices. The Blosum
matrices were constructed using distantly-related sequences. If
you need a highly sensitive search, use a low-numbered Blosum
matrix. The PAM matrices were constructed from a set of
closely-related sequences. For a high-sensitivity search, use
PAM250, or PAM250 Gonnet (based on a more recent dataset.). For
closely-related sequences, use PAM120.
More information of scoring matrices can be found in
Pearson WR (2013)
Selecting the Right Similarity-Scoring Matrix. Curr Protoc Bioinformatics. 2013; 43:
3.5.1-3.5.9.
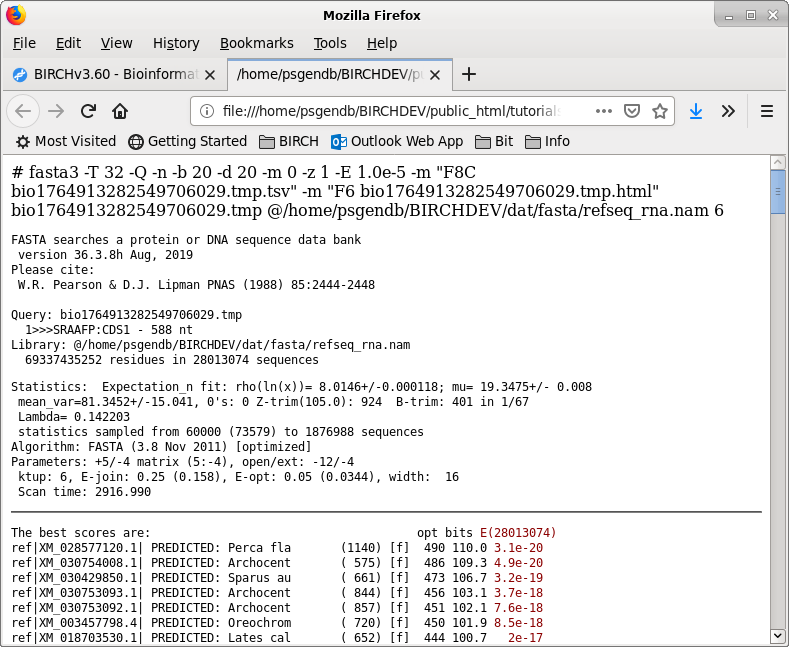
The output from this run can be found in SRAAFP.RefSeqPro.fasta.html
and SRAAFP.RefSeqPro.fasta.tsv.
| Hint: Oligopeptides need
logarithmic score weighting For very short query sequences such as oligopeptides, unweighted scores would not exceed the minimum cutoff values needed to appear in the output. The FASTA programs allow you to specify a logarithmic weighting ratio that gives short query sequences higher matches. The score is weighted by the natural log of the query length divided by the natural log of the database size. |
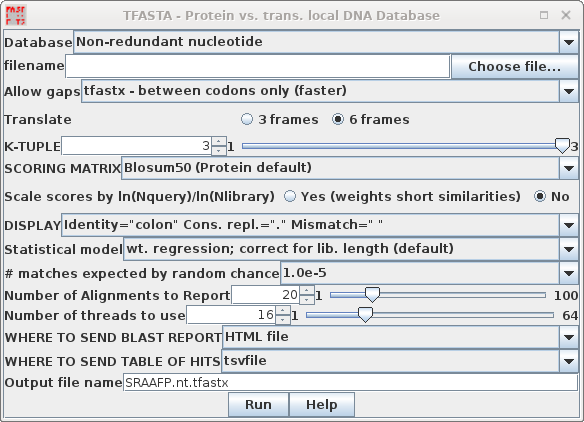
| TFASTA is the most sensitive method for
searching DNA sequence databases. Each DNA sequence is
translated on the fly in either 3 or 6 reading frames (ie.
one or both strands) . ALL of the sequence is translated,
whether coding or non-coding, intron or exon. By default, TFASTA is set to search the GenBank Non-redundant nucleotide database. |
 |
|
Two versions of TFASTA are available. TFASTX allows 3-base insertions (ie. 1 codon), while TFASTY allows 1 or 2 base insertions. TFASTY is therefore the most sensitive program, but it is also slower. TFASTY would be especially good at detecting similarities between a query protein and ESTs, since ESTs typically have more frameshifts than most sequences. |
 |
| Note
on automatic translation: Programs that translate sequences on the fly (eg. TFASTX, TBLASTN, TBLASTX) have no knowledge whatsoever about gene structure (ie. exons, introns, 5' UTR, 3'UTR). All these programs do is take every group of 3 nucleotides and assign a codon to it. Even stop codons are represented by an asterisk (*). Consequently, non-coding sequences and non-coding open reading frames are translated into meaningless amino acid sequences. |
Note: This search took 208 min. of CPU time and about 30 min. of clock time using 32 cores on an AMD Opteron Linux system with 64 Gb of memory, with many sessions running by other users. Be prepared for a bit of a wait. This is primarily due to the size of the nt database. These results will vary greatly, depending on the user load on each system at a given time.
| Hint:
Send
output
from long-running searches to a file.
|
| FASTX and FASTY are the converse of TFASTX
and TFASTY, in that they translate a DNA query sequence,
allowing either codon-sized gaps (TFASTX) or 1 or 2 base
gaps (TFASTY). TFASTY is therefore the slower, but more
sensitive. These programs are particularly well-suited for
comparing a sequence that may have frameshifts (eg. an EST)
with a protein database. To speed things up in this
demonstration, we'll search the RefSeq Protein database. |
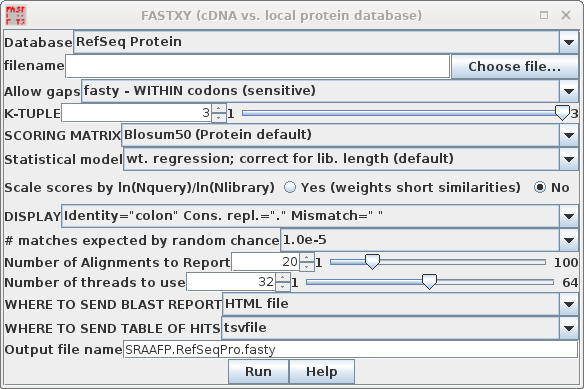 |
Again, since this search will take more time because of fasty,
output goes to SRAAFP.RefSeqPro.fasty.html
and SRAAFP.RefSeqPro.fasty.tsv.
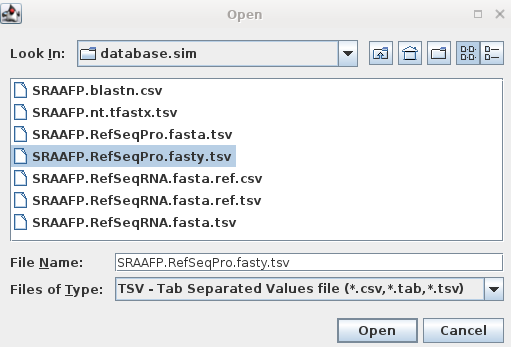
| For example, to retrieve protein sequence
hits from the fasty search in above, start blpfetch at the
command line by typing blpfetch. Read in the names from SRAAFP.RefSeqPro.fasty.tsv using File --> Import Data from TSV file. |
 |
| Since this is a big list, we'll limit the retrieval to the top 20 hits, and send the output to blprotein. Select the first 20 accession numbers by clicking on the cell in the first row, holding down the SHIFT key, and then clicking on the 20 th accession number. |
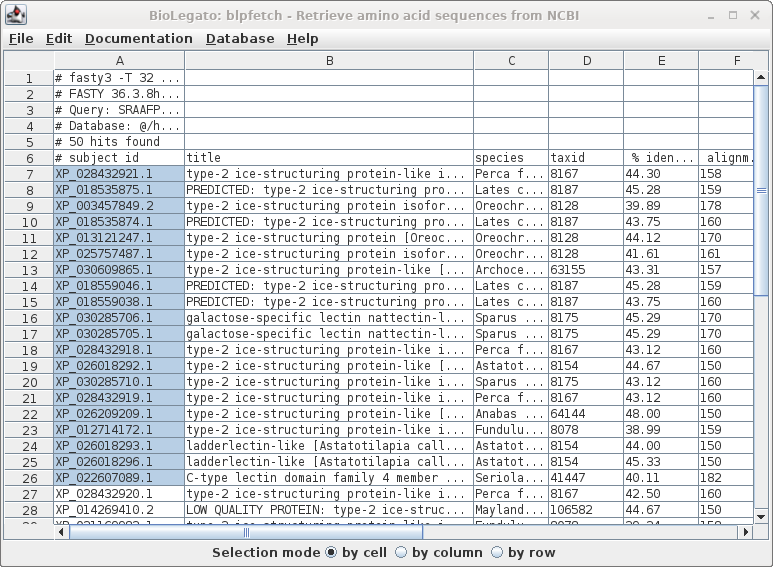
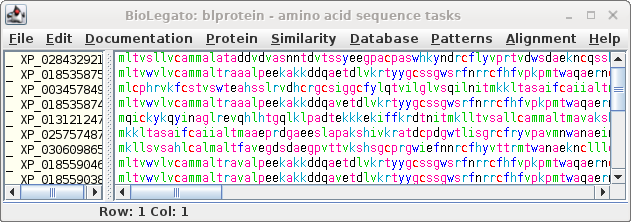
| Next, choose Database --> SeqFetch. We'll
retrieve output in GenBank format, but sent to blprotein so
that we can immediately begin working with the sequences. |
 |
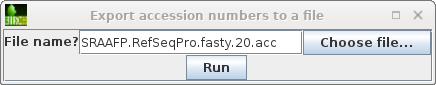
| NCBI Batch Entrex needs a list of Accession
numbers, one per line. We can create this list by selecting
the hits we want, and then choosing File --> Export
accession numbers to a file.Choose a meaningful
filename, and click on Run to save. |
 |
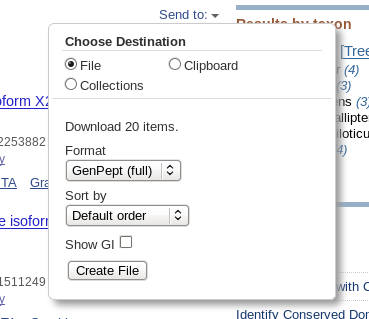
| Click on Send to and fill in the
following: Destination: File Format: GenPept When you click on Create File, a file chooser will pop up. |
 |
| The default name will be something like
"sequence.gb", but it is good practice to give it a name
consistent with the rest of the files in this exercise. A
good name would be SRAAFP.RefSeqPro.fasty.20.gen NOTE: Remember to save this file in the current working directory! Your file chooser will show a different location from the one shown a right. |
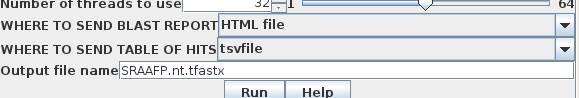 bldna runs all database
searches in the background, ie. as independent jobs. If
you quit bldna or blprotein or even logout, the job will
run to completion. For long-running searches, it is
usually best to send output directly to a file. Simply
select 'Output file', and type an Output file name. In
the example above, output will be directed to two files:
bldna runs all database
searches in the background, ie. as independent jobs. If
you quit bldna or blprotein or even logout, the job will
run to completion. For long-running searches, it is
usually best to send output directly to a file. Simply
select 'Output file', and type an Output file name. In
the example above, output will be directed to two files: