TUTORIAL: DOT-MATRIX SIMILARITY COMPARISONS |
Sept. 29, 2018 |
TUTORIAL: DOT-MATRIX SIMILARITY COMPARISONS |
Sept. 29, 2018 |
DXHOM documentation: $doc/fsap/hom.txt
total 58
-rw------- 1 frist drr 5404 Nov 6 17:22 X52331.gen
-rw------- 1 frist drr 4494 Nov 6 17:21 GMCAB2.gen
-rw------- 1 frist drr 4238 Nov 6 17:21 GMCAB3.gen
-rw------- 1 frist drr 4338 Nov 6 17:29 KPLACBG.gen
-rw------- 1 frist drr 3674 Nov 6 17:21 PEACAB15.gen
-rw------- 1 frist drr 3757 Nov 6 17:22 WHTCAB.gen
Launch bldna, and read in the GenBank files using File --> Open.
Hint:
Similarity comparisons require you to select two or more
sequences at a time.
|
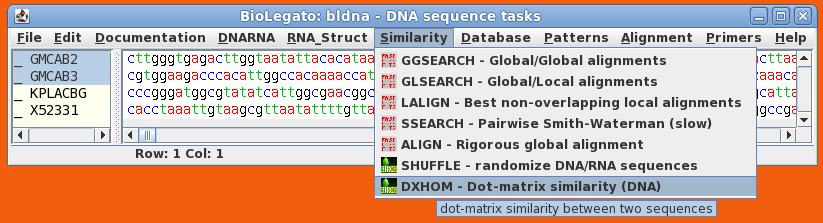
Select GMCAB2 and GMCAB3 by dragging the cursor across the two names, and choose Similarity --> DXHOM. The DXHOM menu will appear:
Output from GMCAB2 vs. GMCAB3
using DXHOM defaults.
a. Compression: Zooming in and out - Each row and column in the matrix represents one or more nucleotide/amino acid positions. By default, A compression factor of 10 is used, with 70 charcters per line, meaning that 700 nucleotides can be represented per line or column. In the example above, the X-axis sequence GMCAB2 is 1354 bp long, so DXHOM must break the output up into two pages, one in which GMCAB2 positons 1..700 are compared with all of GMCAB3, and another in which 701..1354 are compared with GMCAB3. The entire comparison can be fit into a single page by changing the compression factor to 20. [Output with "compression factor" (COMPRESS) =20].b. The signal to noise ratio is controlled by local search window size and similarity cut off value.
The parameter "min. % similarity printed (MINPER)" sets the minimum score for a character to be printed in the matrix. The lower the percent match allowed, the higher the sensitivity, but the greater the background noise due to random chance. [Output with min. % similarity printed=50].
The window size is expressed as the distance from the center of the search window. That is , the search window contains d bases on either side of the center of the k-tuple match. So if the distance is 10, a window of 21 bases (ie. 10 on either side of the central nucleotide) is compared. The wider the search window, the lower the probability of a match by random chance. Thus, lower MINPER values require larger search window sizes. The result from above can be "cleaned up" by doubling the window size. [Output with window Dist. from center of window= 20 ie. window size = 41].
Some reduction in background noise can also be gained by using a k-tuple of 4. This will also speed up the search by a factor of 4, but may be less sensitive.
Finally, the signal to noise ratio can also be improved by simply doing the plot at a lower compression. One way is to use a wider output line (scroll left to right to see entire matrix)
Dist. from center of window: 10
MINPER = 50
COMPRESS = 10
width of output line: (LINEWIDTH) = 130
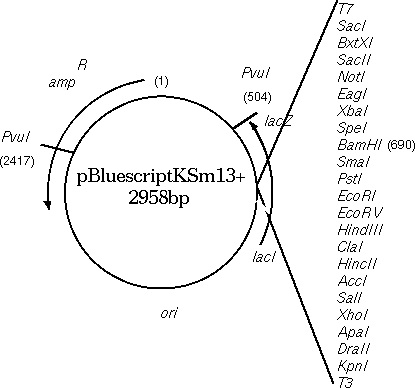
To illustrate the importance of comparing BOTH strands, the
Bluescript vector, containing a beta-lactamase gene for ampicillin
resistance, will be compared with the beta lactamase gene from Klebsiella
pneumonia. Since Bluescript is 2958 bp long, we need to use
COMPRESS= 30 and LINEWIDTH= 100 to make the entire matrix fit onto
one page.
| NOTE: If you
plan to compare both strands of a reference
sequence with one strand of another sequence, in each
plot the reference sequence, or its inverse complement,
MUST be placed on the X-axis.
DXHOM is designed to preserve the coordinate system of
the reference sequence, regardless of which strand is
printed. That is, if you do the first search with the
original strand Bluescript as the reference sequence,
the coordinates of Bluescript would be written left to
right on the X-axis, 1 to 2958. It is INCORRECT to
simply generate the inverse complement of Bluescript and
repeat the search, numbering 1.. 2958. The numbering at
the top should be 2958 downto 1. In that way, a given
coordinate always refers to the same part of the
sequence, regardless of the strand. |
Although there's no straightforward way to get bldna to tell
DXHOM which sequence goes onto the X-axis, when multiple sequences
are selected, they are sent to programs in the order they appear
in the bldna sequence list, top to bottom. Thus, if you want
X52331 to be on the X-axis, it must appear above KPLACBG in the
bldna window. The problem is that because the file chooser reads
files in alphabetical order, X52331 is below KPNLACBG. Select
X52331 and choose Edit --> Cut. Select KPNLACBG, and
choose Edit --> Paste, and ARBLKSP will be pasted above
KPNLACBG.
Compare the original strand of Bluescript (X52331) with the Klebsiella gene (KPLACBG).
Dist. from center of window: 10As seen in the output, there are no significant diagonals, indicating that there are no
min % similarity: 60
COMPRESS=30
LINEWIDTH=100
 Since many people have dificulty
conceptualizing the differences between a strand and its inverse
complement, we'll take a moment to review how these concepts are
defined.
Since many people have dificulty
conceptualizing the differences between a strand and its inverse
complement, we'll take a moment to review how these concepts are
defined.| By default, bldna protects sequences from modification, so those protections need to be unset. Click in the sequence pane next to 'example', and choose Edit --> Get Info.... Uncheck the boxes that say "Ambiguous characters" and "Unambiguous characters". Click on Update. |  |
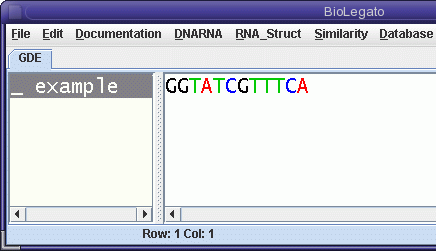
| As shown at right, example
is written 5' to 3' left to right. Select
example, and choose DNARNA
--> blrevcomp. For any sequence, blrevcomp can
create the complementary strand, or flip the strand, or
create the inverse-complement. |
 |
| Let's start by creating the
complementary strand. Choose complement only. |
 |
| Since example is written 5'
to 3' left to right, it should be obvious that example-comp
as displayed in the figure, must be written 3' to 5', left
to right. (Note that blrevcomp automatically adds a suffix to the sequence name to distinguish between the original strand and its diferent transformations.) Next, select example-comp and run blrevcomp again, this time choosing flip (reverse only). |
 |
| Now, example-comp-flip is the
same strand as example-comp, but written 5' to 3' left to
right. |
 |
| blrevcomp can achieve the
same result in one step if you select example and choose reverse-complement in
the blrevcomp menu. As we'd predict, example-opp is identical to example-comp-flip. |
 |
a. Create the inverse complement of X52331.
Select X52331 and choose DNARNA
--> blrevcomp.
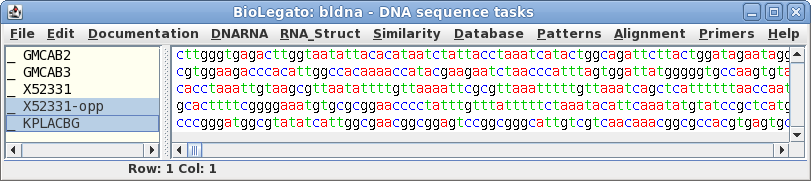
The inverse complement, X52331-opp, is inserted at the bottom of
the bldna window.
When bldna runs DXHOM, it places top-most sequence on the X axis
and the lower sequence on the Y axis. DXHOM is written in such a
way that it can recalculate the coordinates of the X-axis sequence
if that sequence is an inverse complement. For that reason, we
need to move X52331-opp up in the bldna window.
Select X52331-opp and choose Edit
-->
Cut. To insert it below X52331, click on 'X52331'
and choose Edit --> Paste.
b. Compare the inverse-complement of X52331 with KPNLACBG.
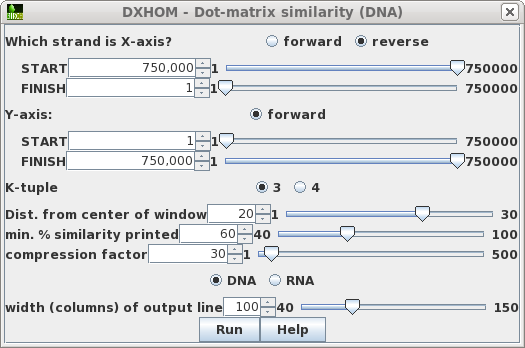
Select ARLKSP-opp and KPNLACBG. Choose Similarity --> DXHOM.| Click on the reverse button to tell
DXHOM that the X-axis strand is the opposite strand. That
will tell DXHOM to write the sequence coordinates in descending
order. This also requires that for the X-axis we set START
at the end of the sequence and FINISH at the beginning of
the sequence. For the X-axis, drag the START slider to the
right, and the FINISH slider to the left. If you don't do this step, the sequence position numbers in the output will be incorrect! |
 |
As illustrated in the map of Bluescript shown above, the
DXHOM output verifies that the opposite strand of Bluescript
has a beta-lactamase gene, going roughly from 2680 downto 1960. [DXHOM output using opposite
strand of X52331].