TUTORIAL:
Basic shell scripting
|
Sept. 28, 2016 |
TUTORIAL:
Basic shell scripting
|
Sept. 28, 2016 |
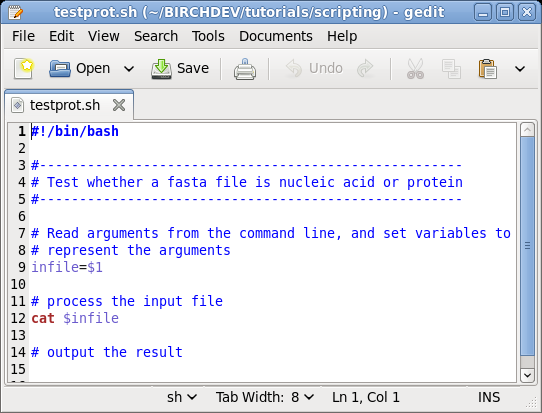
| While you could edit this file in any text
editor, it is best to use an editor that supports syntax
highlighting for various programming languages. On Linux, a good choice is Gedit. At the command line, type 'gedit testprot.sh &' to run the editor in the background. Or, launch Gedit from the Applications menu and open testprot.sh from the File menu. |
 |
eliminate lines beginning with '>' |
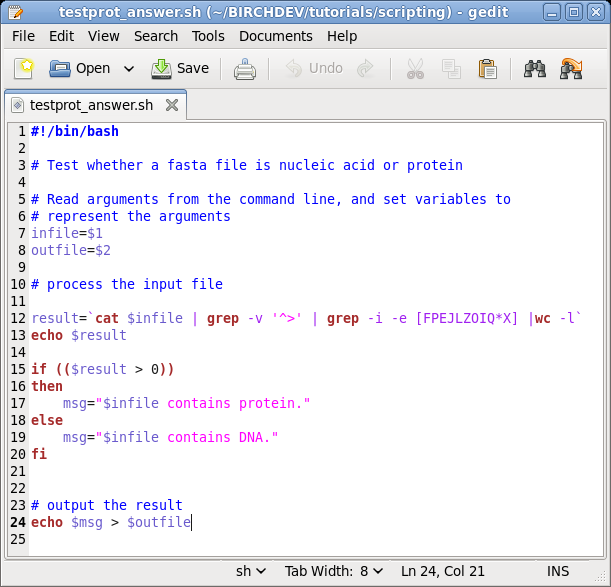
| If you want to see an example of the complete
script, you can look at testprot_answer.sh.
|
 |