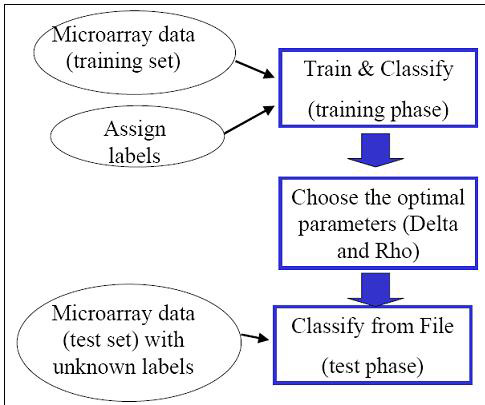
Overview of USC
(Yeung et al 2003)
Prediction of the diagnostic category of a tissue sample from its expression profile and selection of relevant genes for class prediction have important applications in cancer research. We developed the algorithm that is an integrated classification and feature selection algorithms applicable to microarray data with any number of classes.
As with most classification and feature selection algorithms, the USC algorithm proceeds in two phases: the and the phase. A is a microarray dataset consisting of samples for which the classes are known. A is a microarray dataset consisting of samples for which the classes are assumed to be unknown to the algorithm, and the goal is to predict which classes these samples belong to. The first step in classification is to build a “classifier” using the given training set, and the second step is to use the classifier to predict the classes of the test set.
In the training phase, the USC algorithm performs cross validation over a range of parameters (shrinkage threshold Δ and correlation threshold ρ). is a well-established technique used to optimize the parameters or features chosen in a classifier.
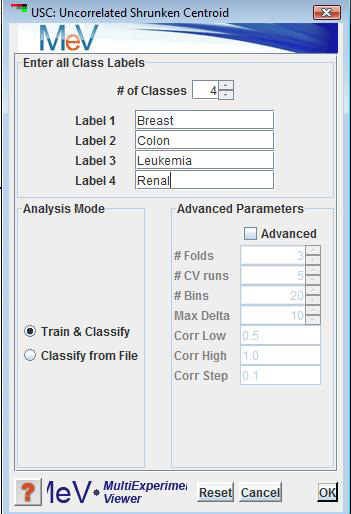
The initial dialog box allows you to choose from 2 modes of operation - or . The option ‘Train & Classify’ should be used for the training phase or if both the training and test sets are uploaded as one microarray data. The option ‘Classify from File’ corresponds to the test phase of the algorithm, and assumes that a classifier has been previously built.
When , the user is required to enter all the unique class labels of the known (training) experiments. By default, there is space for 2 class labels. If more are needed, use the ‘# of Classes’ spinner. ‘Entering Class Labels’ is disabled if you are ‘Classifying from File’.
You are also allowed at this point to make any adjustments to the default parameters. By default, the parameters are disabled. Clicking on the ‘Advanced’ checkbox enables adjustment of the parameters.
: is the number of times to divide the training set in pseudo training and pseudo test sets during a cross validation run. For example: if there are 10 total training experiments to be cross validated and = 5, 2 experiments will be removed as pseudo test experiments during each Cross Validation Fold. After 5 Folds, all 10 experiments will have been used once and only once in the pseudo test set. A higher is recommended for smaller class size.
: is the number of times to repeat cross validation. Reducing this parameter will reduce computation time in the training phase at the expense of less accurate average number of classification errors and genes selected from the cross validation step.
: is the number of different values to use for Delta.
: is the maximum Delta value to use. will range from incrementing by . The user may consider reducing this parameter to get a more precise estimate of the optimal shrinkage threshold Δ if the optimal estimated Δ is significantly smaller than this value. On the other hand, if the number of classification errors from cross validation is unsatisfactory, the user may consider trying a larger .
: is the lowest Correlation Coefficient threshold to use. The default is 0.5, which should be sufficient for most cases.
: is the highest Correlation Coefficient threshold to use. The default is 1.0, which is the maximum possible correlation.
:is the value to increment over going from to .
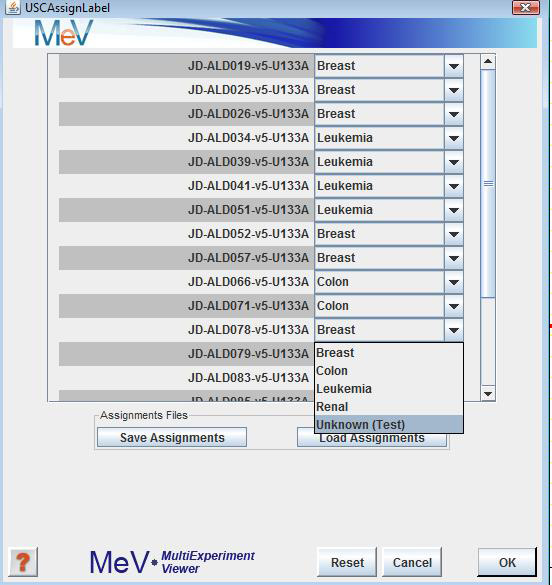
If you are doing , the USC algorithm needs to know the classes of the experiments in the training set. Using the pull down menus, assign labels to each of the experiments that were loaded. Label any test experiments as ‘Unknown (Test)’. Keep in mind that you are not required to test any experiments at this point. You may just classify an entire training set, saving the classifier as a file for later use on any test experiments of choice.
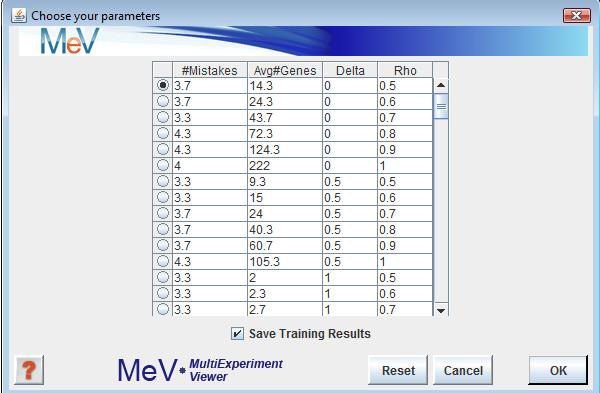
Click ‘OK’ and wait a short eternity for Cross Validation to run. When Cross Validation is finally finished you’ll see the ‘Choose Your Parameters’ dialog box.
During Cross Validation, the USC algorithm has compiled a list of results. During each fold of cross validation, each experiment in the pseudo test set has been tested back against the remaining experiments of the pseudo training set. Here, you’ll be asked to choose between accuracy and the # of genes to use during testing. When the ‘Save Training Results’ checkbox is checked (default), you’ll be prompted to save the training file. If you have any Test experiments, they will be tested now using your chosen Delta Δ and Rho ρ values.
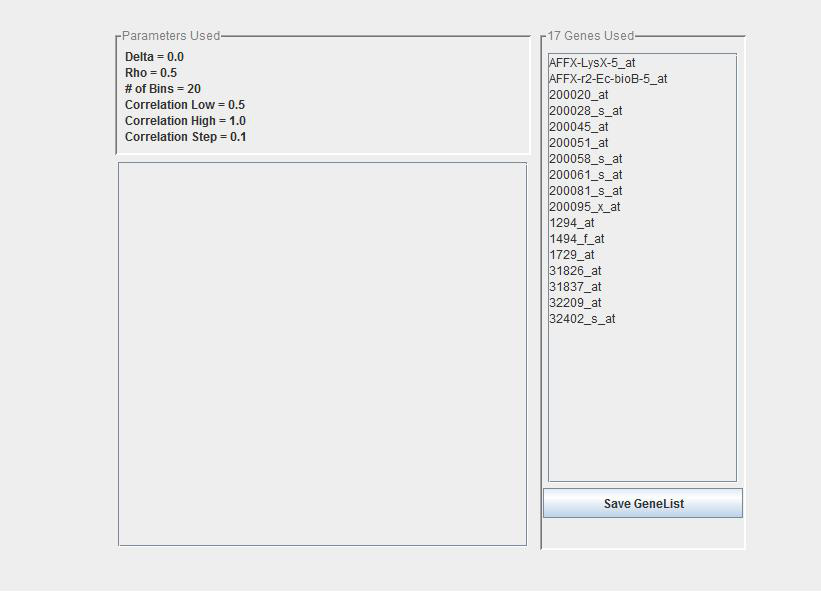
The results of the USC algorithm are returned to the main Analysis Tree in the left pane of the Multiple Array Viewer window. Clicking on ‘Summary’ will display the following view. Any test experiments that were loaded are listed along with their class assignment and the Discriminant score of that assignment. Parameters are also displayed as well as the list of the genes that were used for this classification. You can save that gene list if desired.
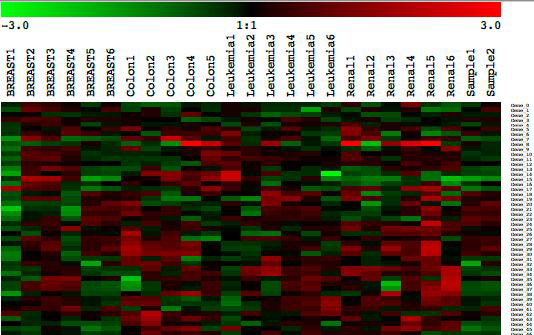
A number of heat map visualizations are also available.
Clicking on displays all the experiments that were loaded and the genes that were used during this classification.
There is also a heat map visualization for each of the classes in the analysis, again, with the genes that were used during this classification.
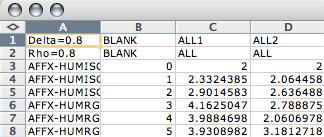
Having saved the results of a classification, you may want to test experiments without the time intensive Cross Validation step. It is important that you use different sets of experimental samples in the training and test phases. Keep in mind that you can only test experiments that are of the exact same chip type as the training experiments.
If you would like to experiment with different values for Delta and Rho, you can easily change them in the Training Result File.