TUTORIAL: SEARCHING LOCAL SEQUENCE DATABASES USING BLAST |
Oct. 5, 2023 |
TUTORIAL: SEARCHING LOCAL SEQUENCE DATABASES USING BLAST |
Oct. 5, 2023 |
| This tutorial assumes that local copies of the Uniprot (Swissprot), NCBI RefSeq RNA and the Non-redundant nucleotide (nt) databases . If local copies of databases are not available, NCBI databases can be searched remotely using NCBI blastp, NCBI blastn etc. |
This tutorial will introduce the several
types of sequence database searches, using the antifreeze protein from the sea raven, Hemitripterus
americanus. The cDNA sequence for this gene is found in SRAAFP.gen.
Create a directory called 'db' and save
this sequence to the db directory.
To read this sequence into bldna, choose 'File --> Open', and select SRAAFP.gen.
|
Since this tutorial will demonstrate database searches
using both DNA and protein sequences as queries, we need
to generate the amino acid sequence. This is done in two
steps. First, we need to extract the protein coding
sequence from the cDNA. (Remember that mRNAs contain both
5' and 3' untranslated sequences. If you look at SRAAFP.gen,
you'll see that the CDS (protein coding sequence) runs
from positions 10 to 597. We will use the FEATURES program
to extract the CDS, and then translate the CDS using
RIBOSOME. |
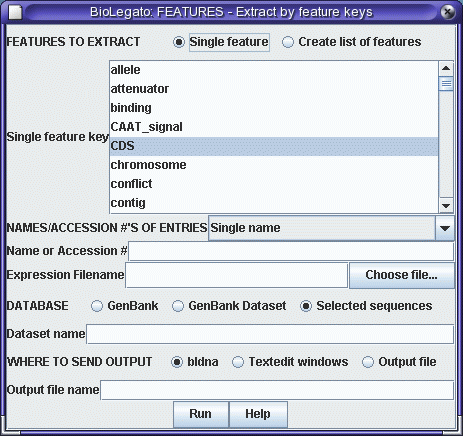 |
Click on 'Run' with the above settings. The CDS will appear in a
new bldna window.
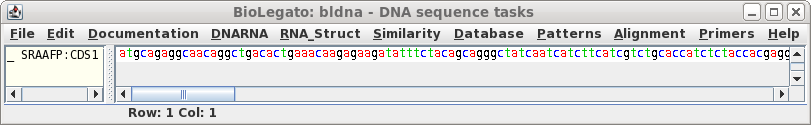
|
Note that the CDS begins with a start codon, indicating
that this cDNA contains the entire protein coding region. Next translate the CDS into protein. Select SRAAFP:CDS1, and choose DNARNA --> Ribosome. By default, Ribosome sends the output to a new blprotein window. |
 |
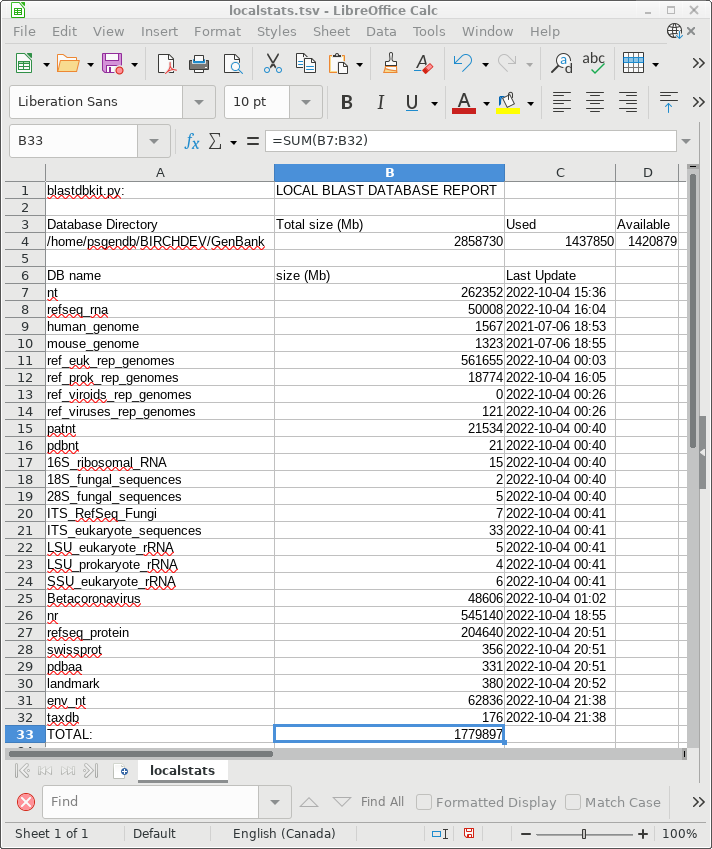
The slowest part of a BLAST database search is simply the process
of reading the data from disk. Therefore, it us useful to have an
idea of how large a database is before running a BLAST search.
BIRCH maintains a list of locally-installed BLAST databases as a
TSV file that can be read by any spreadsheet program. The
spreadsheet can be viewed on the BIRCH web site at Local System
--> BLAST databases. They can also be viewed in bldna or
blprotein in Database --> BLASTDB - report on local BLAST
databases.
| This spreadsheet is meant as a guide for
managing diskspace, but is also useful if we want some idea
of the size of various databases before doing a BLAST
search. Some databases are small, such as the UniProt/Swissprot database (356 Mb). At the other extreme, ref_euk_rep_genomes is about 562 Gb. Aside from the time required to read a database file from disk, BLAST works most efficiently when the entire database can be loaded into memory (RAM). A good rule of thumb is that a database will require roughly half the RAM of the size of the database file on disk. For example, refseq_rna is about 50 Gb, so it would need about 26 Gb of RAM. Where the database is larger than available RAM, data will have to be swapped between memory and disk, which slows the search substantially. |
 |


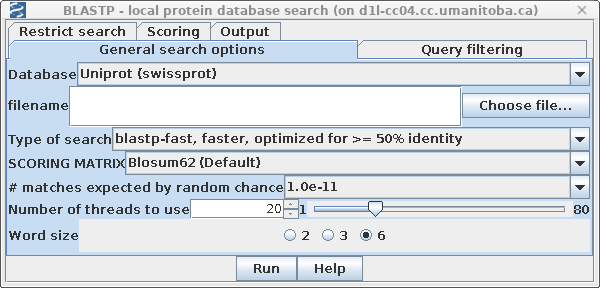
| First, choose a database. The
quickest one to search is UniProt (swissprot), which
contains a small set of carefully-chosen proteins
representative of almost all known proteins. This is
probably the most efficient database for gene annotation or
gene identification ie. which protein does my sequence code
for? |
 |

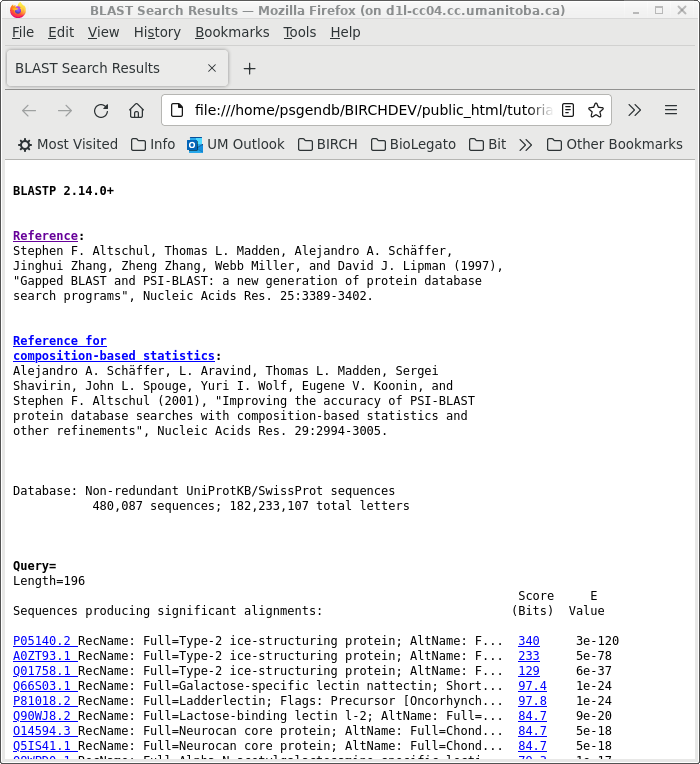
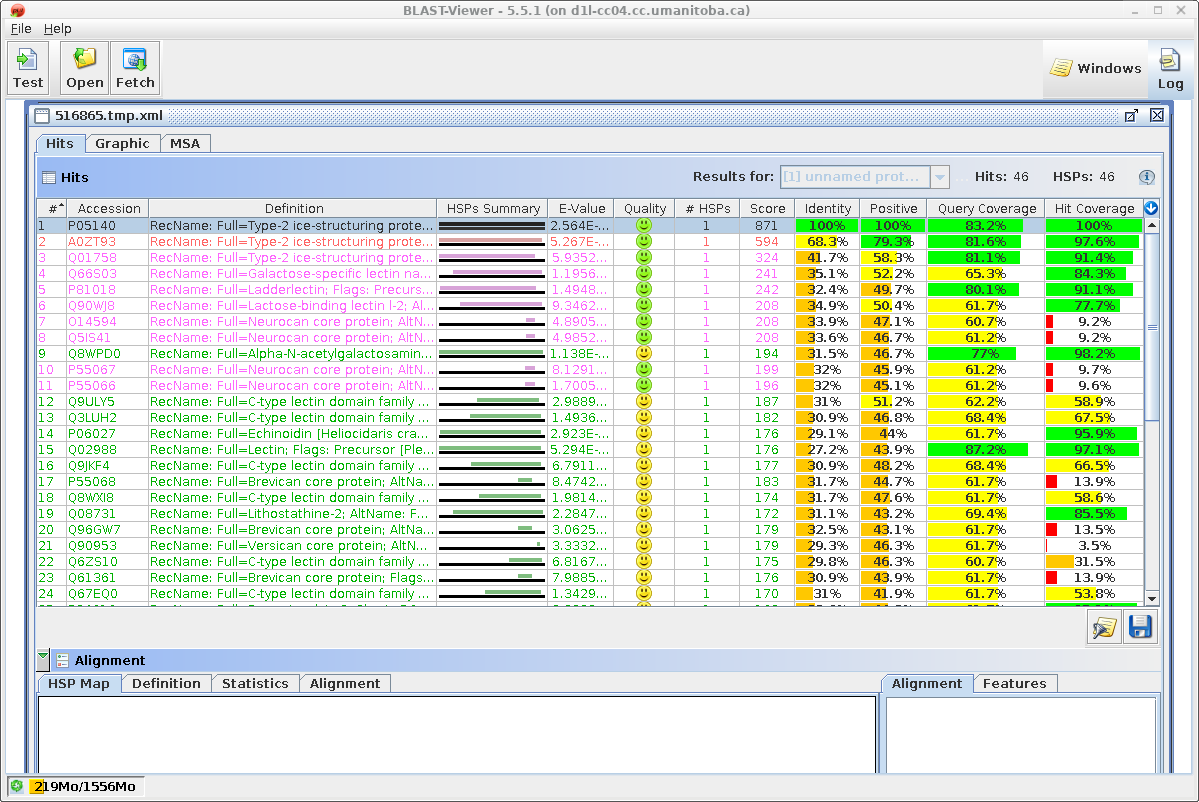
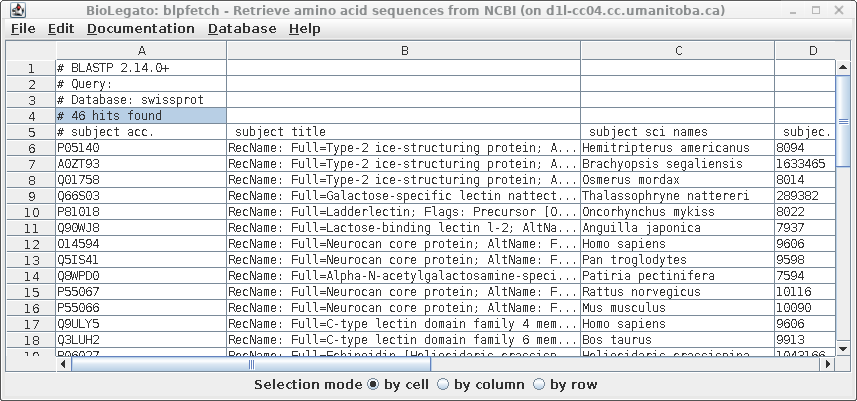
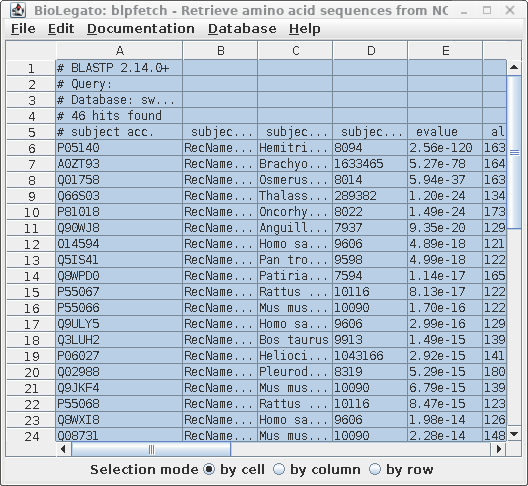
| Similarly, the results in blpfetch can be
saved from blpfetch by choosing File --> Save Selection
As SRAFFP.blastp.swissprot.tsv. |
  |
| A quick word on CSV, TSV and text files,
and BioLegato Most spreadsheet programs can read and write formats called CSV and TSV. CSV files are comma-separated value text files, in which each line in the file represents a row in a spreadsheet. The values for each column in a row are separated by commas. TSV files are TAB-separated value text files. In these files, values in each row are separated by TAB characters. TSV files are probably safer to use, because CSV files make it impossible to include commas within fields. BioLegato programs such as blnfetch, blpfetch and blncbi have a table canvas, that works like a spreadsheet. In these programs, the Open menu can only read text files with a .csv or .tsv file extension. To read files with other extensions eg. .acc, you need to use File --> Import Data from CSV file. Text files such as the one generated above will be read using this approach. |
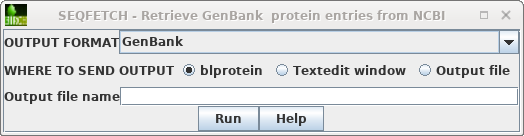
| Retrieving your hits If you decide to retrieve all the hits, this can be done from blnfetch. Choose Edit --> Select All, and then Database --> SeqFetch. By default, SEQFETCH is set to retrieve GenBank entries to a new blprotein window. If you had a large number of entries, it would probably be best to sent output to a file. |
 |
| Hint: Oligopeptides need
logarithmic score weighting For very short query sequences such as oligopeptides, unweighted scores would not exceed the minimum cutoff values needed to appear in the output. The BLAST programs allow you to specify a logarithmic weighting ratio that gives short query sequences higher matches. In the General tab, choose Type of search: blastp, adjust parameters for short queries. The score is weighted by the natural log of the query length divided by the natural log of the database size. |
In some cases your might have a cDNA, a gene fragment or even a
sequencing read. Single reads are more likely to contain errors,
while genomic fragments might contain introns or other non-coding
sequences interspersed with coding sequences. Even if you don't
know the exact translation start and stop sites or the reading
frame, or even the coding strand, it is useful to do a crude
translation of the gene fragment into amino acids. BLASTX simply
takes each group of 3 nucleotides in each of 3 reading frames on
each strand and uses these crude translations as the query.
In this example the query will be the CDS feature that was
extracted from the complete cDNA:
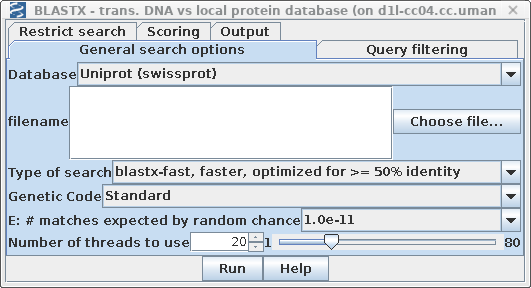
| Select SRAAFP:CDS1, and choose Database
--> BLASTX. Set Database to Uniprot (swissprot). Again this should be a very quick search because the database is small. |
 |
Output
SRAARFP-CDS1.blastx.report.html - BLAST report
SRAAFP-CDS1.blastx.tsv. table of hits
SRAAFP-CDS1.blastx.xml. - XML file for BlastViewer
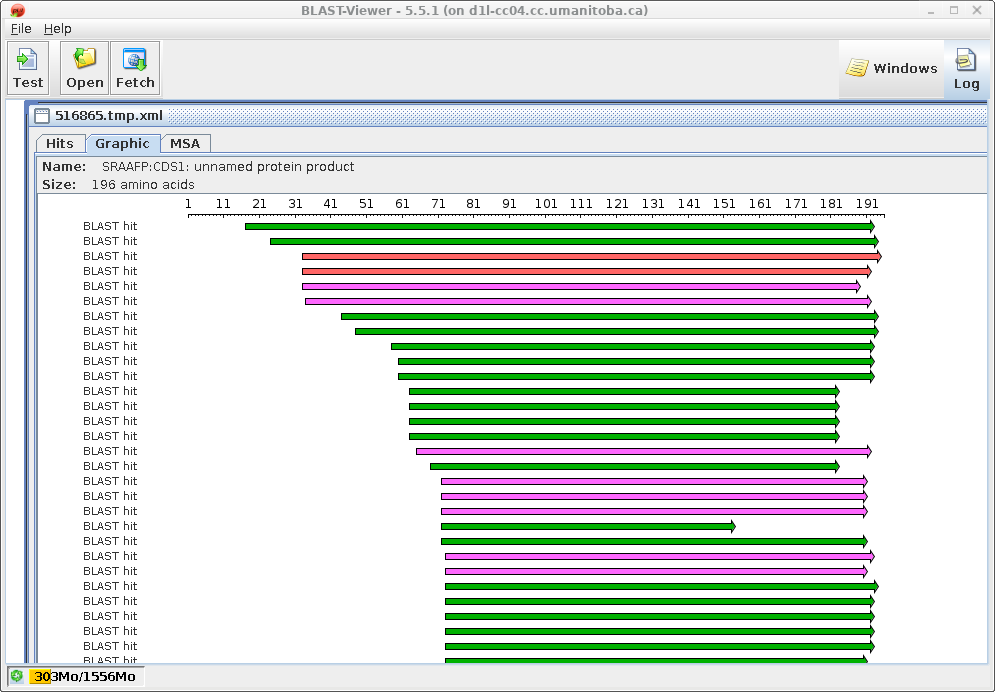
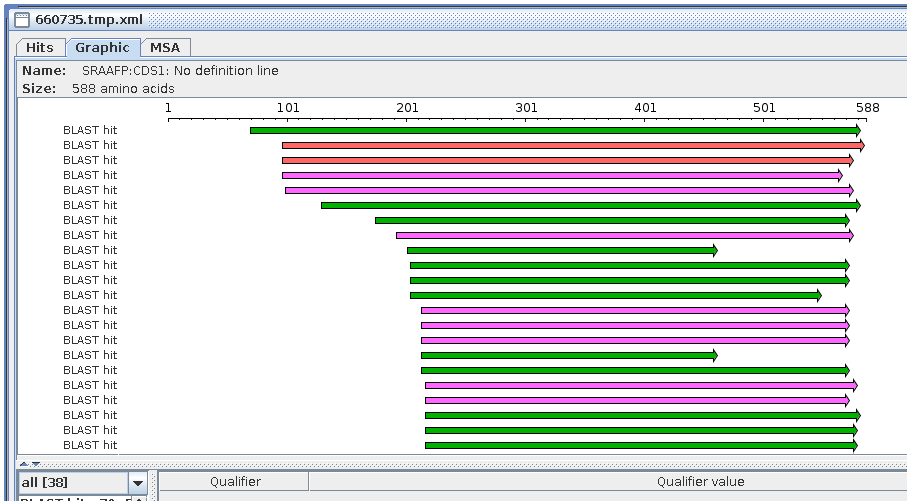
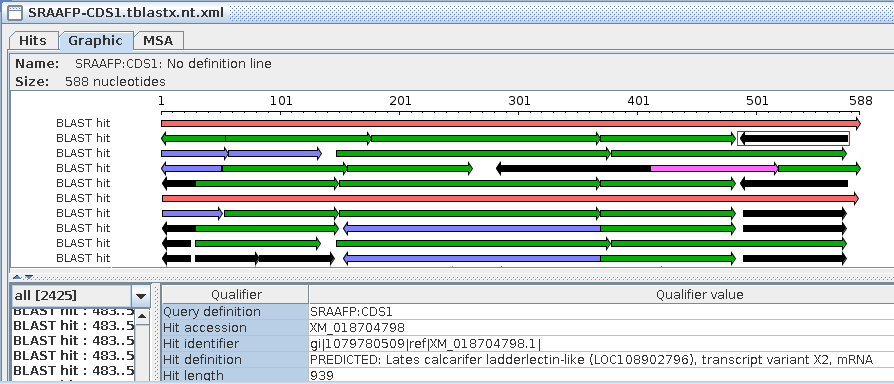
In the BlastViewer output, it is worth noting that the extent of
the matches between query and database sequences is the same as in
the protein vs. protein search. That is, all matches are on the
forward strand (left to right arrows). Many of the hits show
little or no similarity at the N-termini, which is consistent with
the observation that in many protein families, N-terminal
sequences diverge rapidly, while core sequences tend to be more
conserved.
| Note
on automatic translation: Programs that translate sequences on the fly (eg. BLASTX, TBLASTN, TBLASTX) have no knowledge whatsoever about gene structure (ie. exons, introns, 5' UTR, 3'UTR). All these programs do is take every group of 3 nucleotides and assign a codon to it. Even stop codons are represented by an asterisk (*). Consequently, non-coding sequences and non-coding open reading frames are translated into meaningless amino acid sequences. |
In your bldna window, select the amino acid sequence SRAAFP:CDS1, and choose Database -->blastn (DNA vs. DNA database). In order to detect more distantly related sequences, set Type of search to blastn (default is Megablast), and set E: # of matches expected by random chance to 1.0e-5 (stands for 10-5).
|
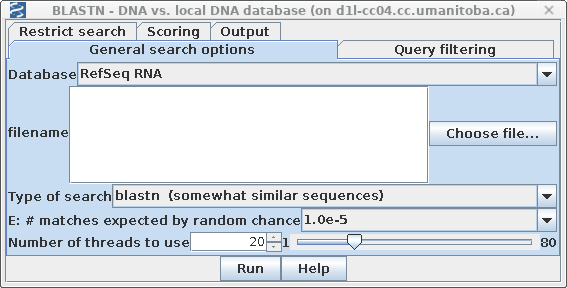 |
| RefSeqRNA
is a fairly large database (~50 Gb at this writing.) Since
the search may take awhile, let's tell BioLegato to send
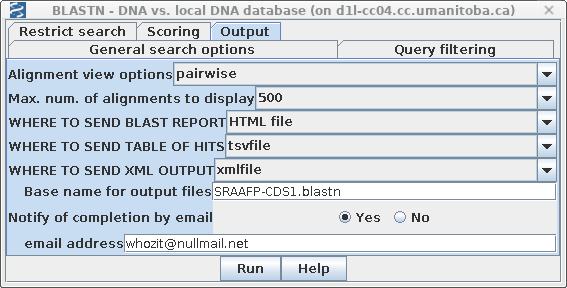
an email notifying us when the search is complete. Consequently, it may be convenient to send output directly to files, rather than to the screen. The search can run in the background, and the user can view the files at a later time. Open the Output tab and set Notify of completion by email to Yes. It is usually most convenient to set the email address to one that will go to your phone or mobile device. We want output to be saved to files, rather than popping up on the screen. Choose HTML file for the destination of the BLAST report, the tsvfile for the table of hits, and xmlfile to create an XML file that could be read by BLASTviewer. Set the base name for these files to SRAAFP-CDS1.blastn.All filenames will begin with this basename, and will get file extensions .html. .tsv and .xml, respectively. |
 |
| How many threads should
I use? BLAST can break the database search into 2 or more threads which work simultaneously on different parts of the problem. The more threads, the faster the search will be completed. Most desktop computers and laptops will have at least 2 or 4 CPUs, while the example at right is taken from Biolegato running on a server with 64 CPUs. In the General menu, BioLegato will set the default number of threads to 1/4 of the number of CPUs available on your computer. If you increase the number of threads, always use at least some free CPUs for other tasks on the system. In a workshop setting where many students are running BLAST searches at the same time, it is best to limit each student to a few threads. Each search will take longer, but that is a trade-off to avoid an overall degradation of system performance. |
 |
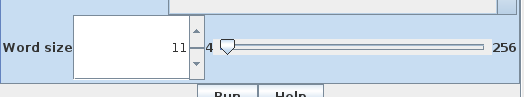
| Hint: For more
distantly-related sequences, use Word size = 4 or 6.
In the Scoring tab, the word size is 11 by default. Setting a smaller word size would provide some improvement in sensitivity, at the expense of speed. It is usually better to allow a larger E value (eg. 1.0e-5) to detect more distantly-related matches. |
 |
Output files:
BLAST Report - SRAAFP-CDS1.blastn.html
table of hits - SRAAFP-CDS1.blastn.tsv
XML file - SRAAFP-CDS1.blastn.xml
| What factors affect
BLAST performance? The BLAST programs are computationally very fast. By far the most important factor affecting performance is the fact that BLAST reads the entire database from disk into memory. When databases reside on an internal disk drive, the database can be read quickly. Solid-state drives have faster read times than magnetic (HDD) drives. On server systems where files are mounted remotely across the network from a file server using NFS, the time needed to read the database can be very long. For example, This search took 37 min. on a using 20 threads on an 80-core Dell R815 Linux server with 1 Tb of memory, with many sessions running by other users. (As an interesting aside, NFS filesystems will cache files on the local machine. That means that once you do one search on a particular database, subsequent searches will be lightening fast, because the database is already cached on the local machine.) RAM also matters. If the size of the database is larger than available memory, BLAST searches may take much longer because the data will need to be swapped between disk and memory. |
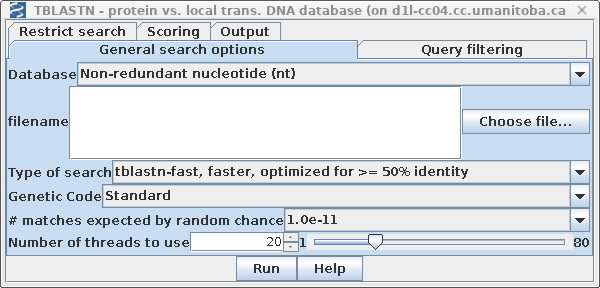
| Each DNA sequence in the nt database is
translated on the fly in either 3 or 6 reading frames (ie.
one or both strands) . ALL of the sequence is translated,
whether coding or non-coding, intron or exon. By default, tblastn is set to search the GenBank Non-redundant nucleotide database. Set E: # matches expected by random chance to 1.0e-11. |
 |
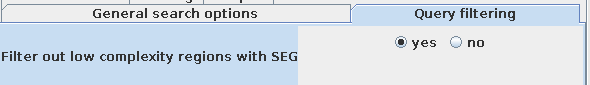
| The nt database contains many genomic
sequences with short tandem repeats, which are referred to
as low complexity regions. Low complexity sequences might
skew the distribution of scores. Therefore, it is best to
turn on filtering in the Query filtering tab, as shown at
right. |
 |
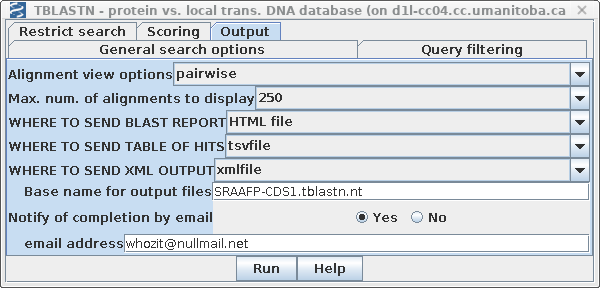
| Since the non-redundant nucleotide database
is quite large (262 Gb at this writing), the search may take
awhile. Consequently, it may be convenient to send output
directly to files, rather than to the screen. The search can
run in the background, and the user can view the files at a
later time. In the Output tab, choose HTML file for the destination of the BLAST report, the tsvfile for the table of hits, and xmlfile to create an XML file that could be read by BLASTviewer. Set the base name for these files to SRAAFP-nt.tblastn. |
 |
BLAST Report - SRAAFP-CDS1.tblastn.nt.html
table of hits - SRAAFP-CDS1.tblastn.nt.tsv
XML file - SRAAFP-CDS1.tblastn.nt.xml
| The main search parameters remain the same. Remember to set Filer out low complexity regions with SEG to "Yes" in the Query filtering tab. |
 |
BLAST Report - SRAAFP-CDS1.tblastx.nt.html
table of hits - SRAAFP-CDS1.tblastx.nt.tsv
XML file - SRAAFP-CDS1.tblastx.nt.xml
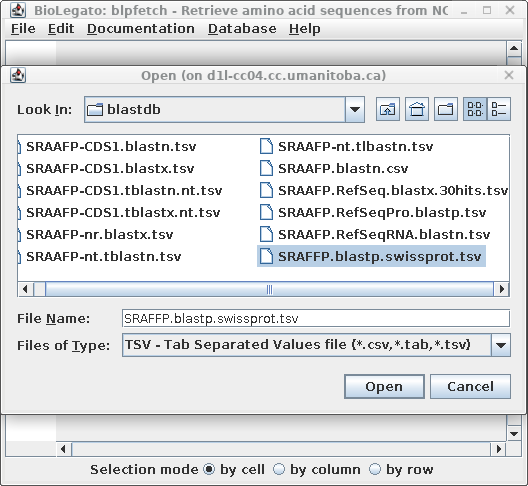
| For example, to retrieve protein sequence
hits from the blastp search in part 3 above, start blpfetch
at the command line by typing blpfetch. Read in the names from SRAFFP.blastp.swissprot.tsv using File --> Open. |
 |
| To retrieve the sequences for these hits,
choose Edit --> Select All, and then Database
--> SEQFETCH. |
| Next, choose Database --> SeqFetch -
Retrieve protein entries from NCBI. By default,
sequences will be retrieved in GenBank format. Note: When there are a large number of sequences you may wish do select some subset of the hits for retrieval. |
 |
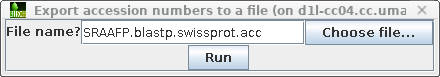
| BatchEntrez
reads a file of accession numbers and returns
sequences. Therefore we first need to save the accession
numbers previously selected to a file.
In blpfetch, make sure the accession
numbers
you want are selected and
then choose File -->
Export accession numbers to a file.
Set an output filename eg. SRAAFP.blastp.swissprot.acc. |
 |
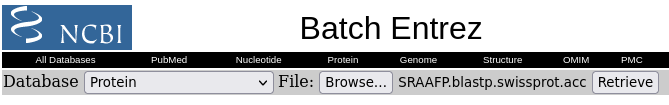
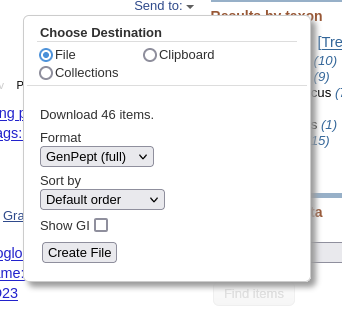
| Click on Send to and fill in the
following: Destination: File Format: GenPept (full) When you click on Create File, a file chooser will pop up. |
 |
| The default name will be something like
"sequence.gp", but it is good practice to give it a name
consistent with the rest of the files in this exercise. A
good name would be SRAAFP.blastp.swissprot.hits.gen NOTE: Remember to save this file in the current working directory! Your file chooser will show a different location from the one shown a right. |