TUTORIAL: Comparing genomes using Mauve |
Nov. 22, 2023 |
TUTORIAL: Comparing genomes using Mauve |
Nov. 22, 2023 |
| References: Mauve Web Site This tutorial continues from the previous tutorial Comparing genomes using dotplots. |
Note: The current version
of Mauve (2015-12-13) requires Java8. On some systems Java11
is the default. If Mauve fails to launch you may need to install
OpenJDK8 on your system. |
| sshcc |
login to a compute node.
sshcc will automatically choose a machine with the lowest
load average. You should see the command prompt change to indicate which machine you are logged into (eg. cc07). Keep in mind that while this terminal window is now on a cc node, the rest of your desktop is still running on the original login host. |
| cd tutorials/getgenome | When you login to a new machine using ssh, you start out in your $HOME directory. Therefore, we need to go to the getgenome directory to work in that directory. |
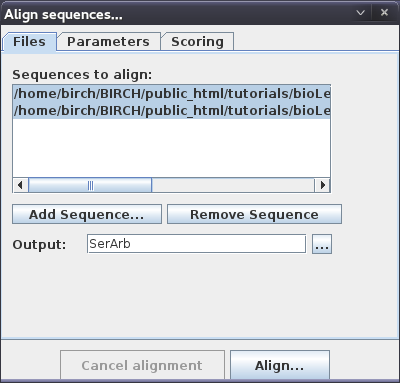
| You can add as many genomes as you wish in
the Align sequences window. For now, read in GCA_000146045.2_R64_genomic.fna GCA_000292725.1_SacArb1.0_genomic.fna We'll use the Output name "SerArb" for this comparison. Click the Align button to begin the alignment. |
 |
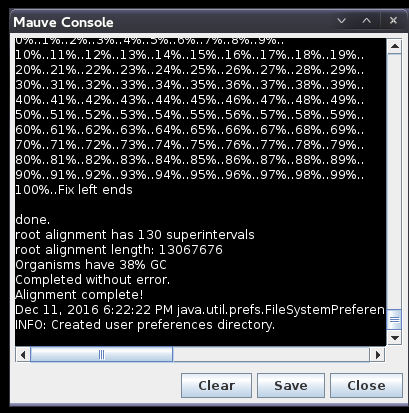
| Progress
of the alignment is shown in a popup window. This
alignment will probably take several minutes. The window will indicate "Done" when the alignment is complete. After a slight delay, the alignment will appear in a new Mauve window. |
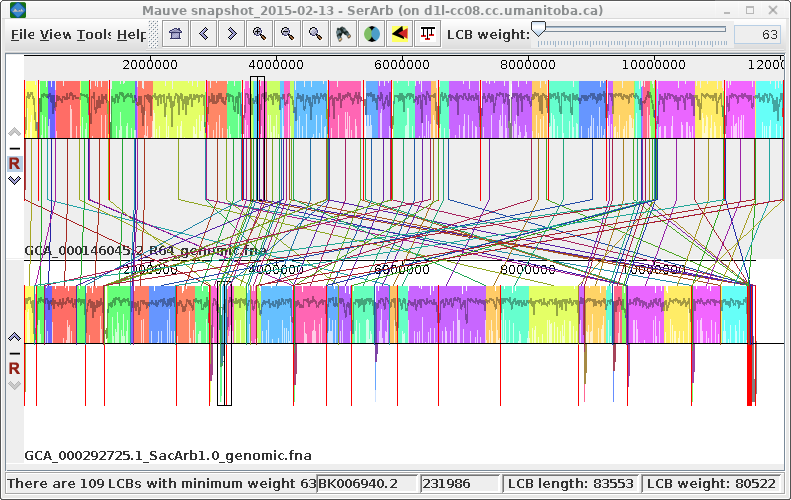
 |



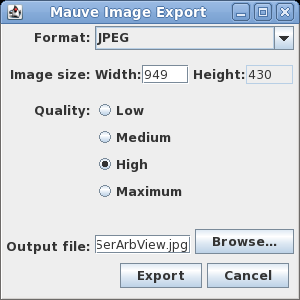
| Exporting images - You can
export the current view of the alignment to an image file
by choosing Tools --> Export Image. In the example at right, the default format is JPEG, and the image is saved to a file called SerArbView.jpg. |
 |
| Resuming a Mauve session - When
creating the alignment, Mauve saves all files related to
the alignment in the current working directory. In the
example, we ran Mauve with the Output name 'SerArb'. All
files will begin with SerArb as the basename eg.
SerArb.guide_tree. You can quit Mauve and resume your
session by choosing File --> Open Alignment. Click on
the file named SerArb (with no extension) and the
alignment files will be read back into Mauve. |