TUTORIAL: VISUALIZATION
OF PHYLOGENETIC TREES
|
June 25, 2021 |
TUTORIAL: VISUALIZATION
OF PHYLOGENETIC TREES
|
June 25, 2021 |
| This tutorial continues from the previous tutorial Phylogenetic
Analysis Using Distance Methods. It begins with
multiply aligned protein and DNA sequences for plant type
III chitinases. To eliminate gappy positions, the alignments
were processed by Gblocks. The starting point for this
tutorial is the Gblocks output alignments. Purpose: To demonstrate the power of tree visualization using Archaeopteryx. Archaeopteryx is very feature-rich, so we will only demonstrate some of its capabilities here. |
| The phyloxml files contain only the Accession
numbers of the sequences, the tree topologies, branch
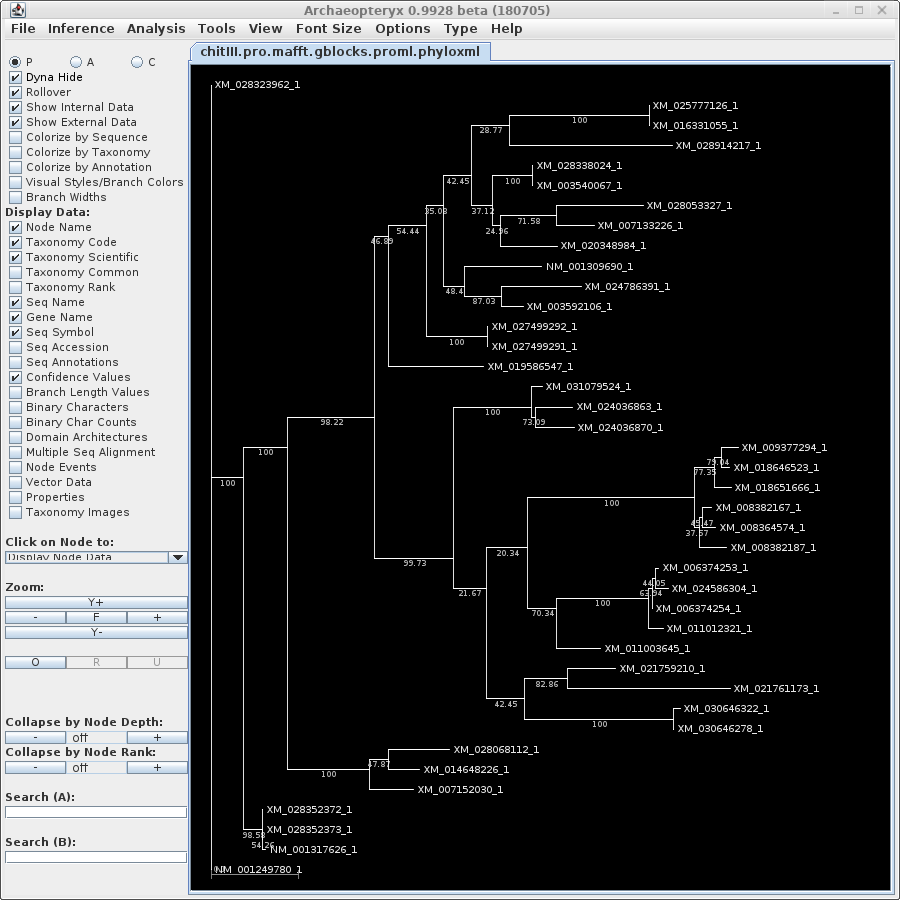
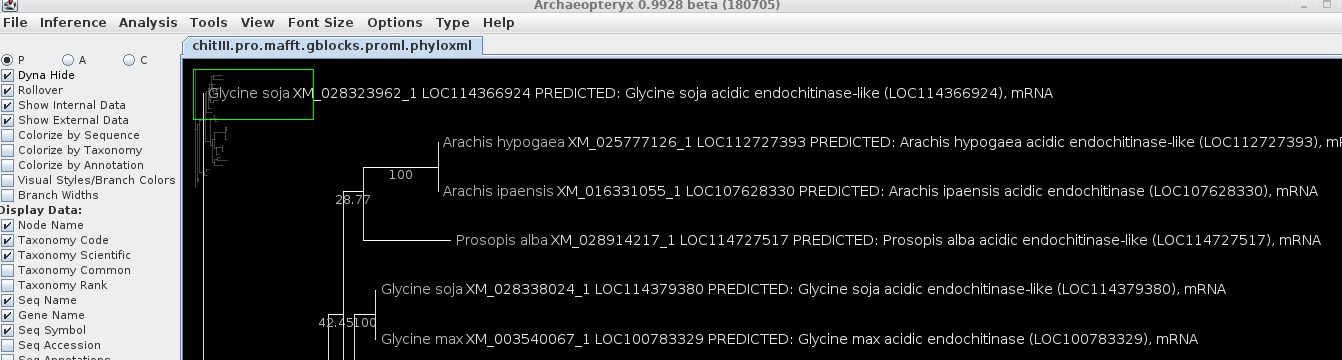
lengths, and bootstrap replicate numbers. cd into the trees directory and launch blnalign. Launch Archaeopteryx either by typing 'archaeopteryx' at the command line, or from BIRCH --> Phylogeny. In this case, we'd like to know the species from which each sequence is derived. One of the great features of Archaeopteryx is the ability to retrieve this information from databases, using the accession numbers of the sequences in the tree. |
 |
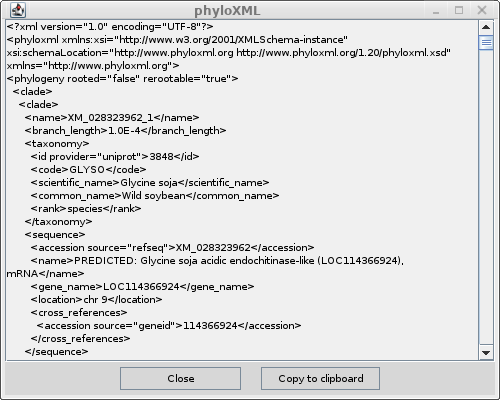
| It is instructive to have a look at the XML
code by choosing View --> as phyloxml. If you have ever made web pages, you'll be familiar with XML. HTML is a subset of the more general XML format. For each type of data, there is an XML specification. So HTML is one kind of XML, phyloxml is an XML for specifying phylogenetic trees and associated metadata. The formal definition can be found at phyloxml.org. |
 |
| protein |
DNA |
 |
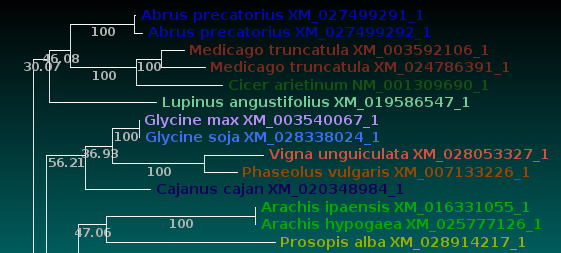 |
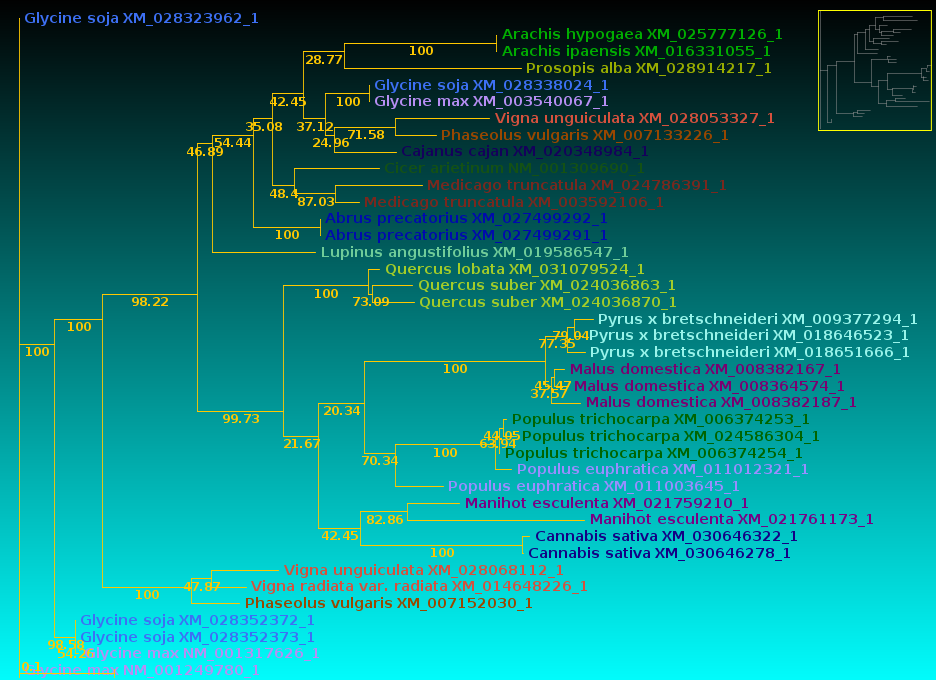
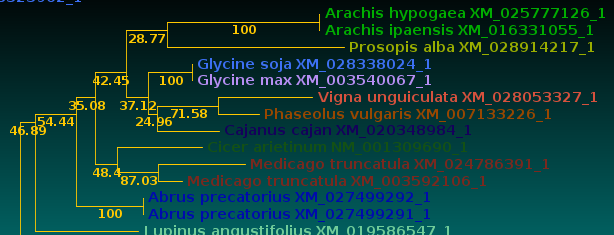
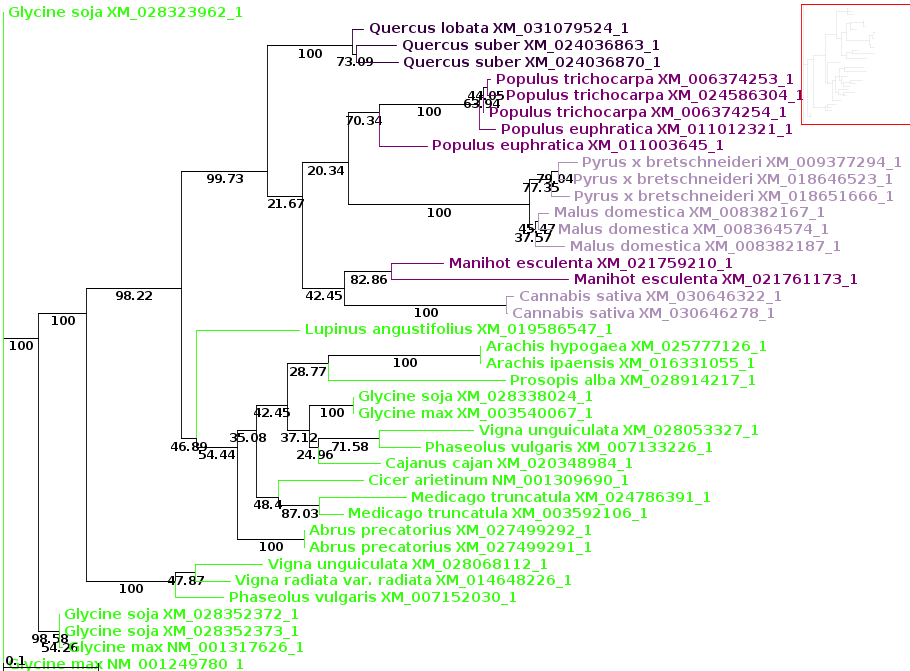
| The Click on Node to: tool on the control
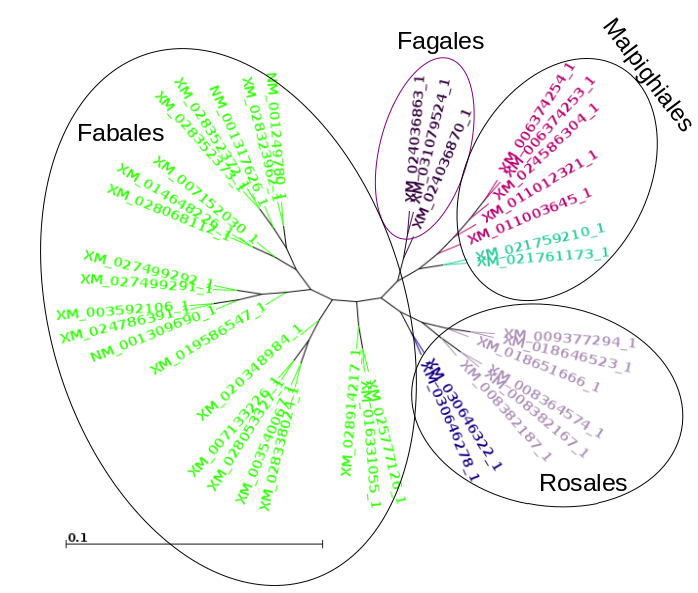
panel. As a trivial example, the protein tree shows the Glycine soja sequence above the Glycine max, while the DNA tree show Glycine max above Glycine soja. To eliminate this meaningless difference between the two trees, click on the outer node joining the two Glycine sequences in the DNA tree: 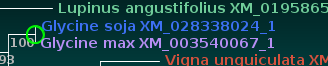 The green circle shows where the node was clicked to swap the branches. |
 |
| protein |
DNA |
 |
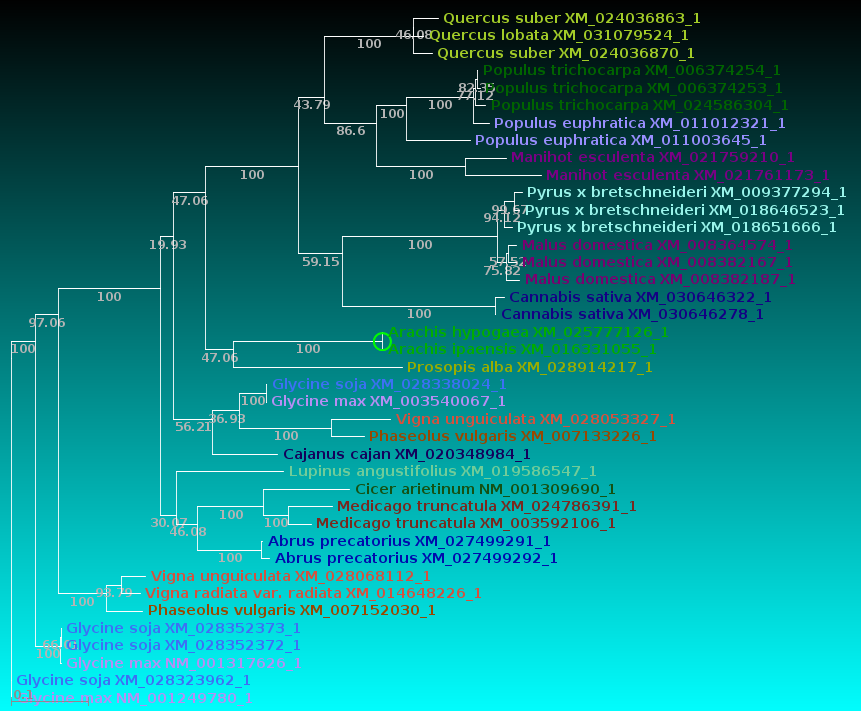 |
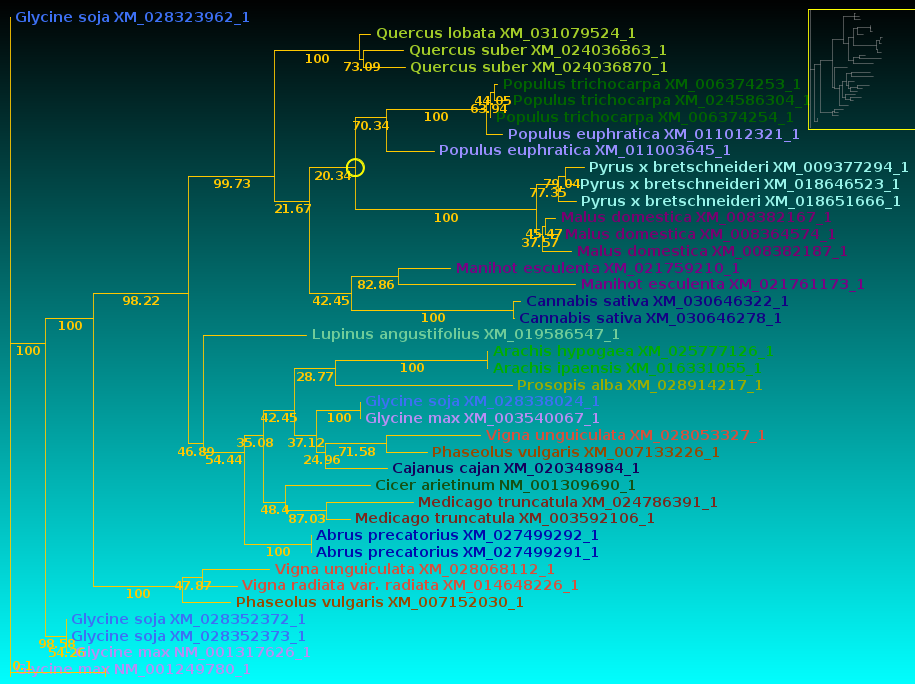
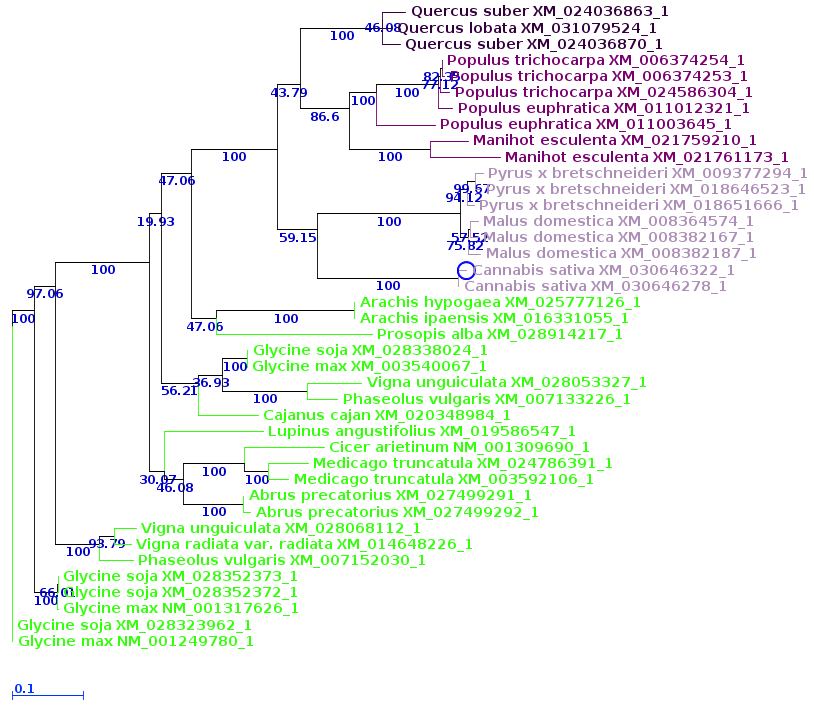
| Now, let's compare how the protein and DNA
trees separate species
based on a higher taxonomic rank. For both protein and DNA trees, Tools --> Colorize subtrees by taxonomic rank, and choose "Family". The most striking thing shown in the comparison is that while the protein alignment groups the sequences from Manihot (Cassava, order Malpighiales) with Pyrus, Malus and Cannabis (order Rosales), the DNA tree groups Manihot with Populus, which is also in Malpighiales. Bootstrap values indicate that Populus and Manihot were together on 86.6% of replicate trees. |
|
| protein |
DNA |
 |
 |