Lecture 9, part 4 of 4
| PLNT4610/PLNT7690
Bioinformatics Lecture 9, part 4 of 4 |
Units of Data
| Unit |
Value |
Example |
| Megabyte (Mb) |
106 bytes |
prokaryotic genome ~ 106 - 107
bp |
| Gibabyte (Gb) |
109 bytes | human genome = 3 x 109 bp total length of reads to sequence human genome = 1.4 x 1011 nt combined nucleotide sequences in NCBI GenBank 245.0 = 1.4 x 1014 bp |
| Terabyte (Tb) |
1012 bytes | Daily output of 157 DNA sequencers at
Beijing Genomics Institute1 = 6 x 1012
nt |
| Petabyte (Pb) |
1015 bytes | 2013 European Bioinformatics Institute
Databases = 2.0 x 1016 bytes Annual data output of Large Hadron Collider = 1.5 x 1016 bytes Library of Congress, including multimedia = 2.0 x 1016 bytes |
| Exabyte (Eb) |
1018 bytes | All
words ever spoken by human beings (as written
text) = 5 x 1018 bytes 2013 Estimate of Google disk storage = 1.0 x 1019 bytes |
| 1 Marx, V (2013)
Biology: The big challenges of big data. Nature
498:255-260. doi:10.1038/498255a 2 Exabyte - Wikipedia https://en.wikipedia.org/wiki/Exabyte |
||
RAM - Random
Access Memory - All data used in computation resides in
RAM. To work on data from a disk, it is necessary for a program
to read a copy of the data into RAM.
CPU and core - A
Central Processing Unit performs operations on data in RAM.
Originally, the CPU had one processor. Today, the vast majority
of CPUs manufactured today, even on low-end PCs, have 2 or
more cores, each of which can process information independently.
The terms CPU and core, while not synonymous, are often used
interchangable. Strictly speaking, it is most correct to use the
term core to refer to the number of processing units.
GPU - "Graphics
Processing Unit" - Originally developed for rendering graphics
and applications such as gaming, many types of processing can be
accelerated, rather than CPUs.
compute node -
An individual computer belonging to a cluster. Can also refer to
almost any computer on a cloud.
cluster - a
group of computers that functions as a single computer, often
sharing memory
Shared memory architecture - A system in which each processing unit, by way of a core processor for example, can access the entire memory space. This is typically the case with a traditional computer, where two processes can easily share the memory to exchange information quickly.
MPI - Message Passing Interface - A communication standard for nodes that run parallel programs on distributed memory systems such as HPC clusters.
cloud - Not really an HPC term per se. The cloud refers to a large array of computers that can provide computing capability as needed. While the cloud does not imply HPC capabilities, HPC systems can be offered as part of a cloud.
Serial computing
- Working on a problem step by step until it is complete, on a
single CPU. Most computer programs fall into this category.
Parallel
computing - The art of breaking large computational
problems into smaller problems that can each be solved
simultaneously by a large number of CPUs. Parallel programming
requires specialized strategies for re-thinking computational
problems so that they better lend themselves to parallelization.
Some languages such as C++ and Fortran have extensive libraries
that handle common tasks in parallel computing.
| What does a petabyte
look like? See the table full of hard drives needed for NASA to image a black hole in Whitwam R (2019) It Took Half a Ton of Hard Drives to Store the Black Hole Image Data. Extreme Tech, April 11, 2019. It was quicker to fly those disk drives from the Hawaii Mauna Kea Observatory to MIT in Boston than it would have taken to send the data over the Internet. |
A virtual machine
implements the instruction set of a computer (eg. Intel, AMD
chip) in software. Effectively, it behaves identically to a
physical computer. Any operating system can be installed on a
virtual machine, and it will boot exactly the same as if it was
a physical machine. Virtual devices, such as hard drives, memory
and CPUs map to real components of the computer on which the VM
is running. Each VM is thus guaranteed a certain amount of
resources. One advantage is that resources can be reallocated
dynamically. The down side is that the user ONLY has access to
the allocated resources, and not to the full resources of the
real machine.
|
|
Should
I buy a server for my lab/department/institute, or use
VMs?
| Advantages |
Disadvantages |
|
| System administration |
If bioinformatics software is pre-configured,
you get started very quickly. |
You still need to install any bioinformatics
software that didn't come pre-installed. You still have all the system administration work required for a stand-alone Linux system. If you already have a Linux server, running a VM on the server makes very little sense. It is more work, not less. |
| Resources |
You can expand or shrink the amount of RAM or
CPUs as needed, so you only pay for resources you need, and
time you spend on the VM. |
Default VM configurations are no bigger than
what you have on a desktop PC eg. 16 Gb RAM, 4-8 CPUs. To
expand those resources costs money. If you are using hundreds of Gb or Terabyte amounts of data, disk space on a VM can be very expensive. On your own computer, you can buy disk space very cheaply! |
| Backups |
Vendor will (might or might not?) backup up
your data. If you own a server, it's up to you to buy Network-Attached Storage or storage on the Cloud, and to configure your own backups. |
|
| Cost |
VMs are potentially a lot cheaper than buying
a server and maintaining the hardware. The problem of
upgrading hardware is up to the host. |
VMs can also be very expensive if you keep
them running 24/7 (ie. a virtual server). As your number of
users increase, the ongoing cost increases. |
| Performance and availability |
If your private server goes down, somebody
has to be ready to fix the problem. The vendor who provides the VM will have enormous redundancy, so there will always be compute nodes and CPUs available. |
You can only get at your data while the VM is
running. Anything running on a VM will run a bit more slowly than "software on metal". However, the performance of VMs is actually very good. |
There is nothing
particularly special about VMs. They are simply one more tool in
the tool kit. Like any tool, there are good use cases, and also
cases where a VM makes no sense.
One thing VMs are
really good for: testing software
If you produce any sort
of software that you want to distribute to other users, it is
important to test the software on as many platforms as possible.
Instead of buying a separate computer for each platform, you can
create as many VMs as you want on a single machine, each running
a different operating system, version, or configuration of
virtual hardware.
Guideline:
Unless you have a compelling case for a VM, just use a real
machine.
a) Running many
serial jobs at the same time - You don't need to be an
expert in HPC to take advantage of HPC capabilities. A crude
form of parallel computing can be done in case where you know
that a problem can easily be divided into many parts that can be
done simultaneously. Simply break the dataset into as many parts
as you have cores, and launch them at the same time. The
operating system will take care of assigning each instance of
the task to a core, and the net effect will be parallel
computing. With a little knowledge of programming, you could
write a script that breaks up the dataset, runs the jobs, and
combines the output into a single file when all jobs have been
completed.
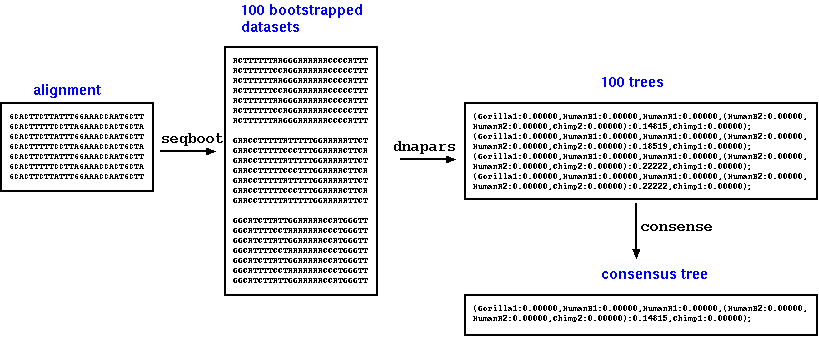
Normally, to construct
a parsimony tree from bootstrap replicates, you would create a
file containing your bootstrapped datasets (eg. 100 replicates)
by running a single instance of seqboot. Next you would run
dnapars to make trees from each dataset, and then consense to
create a consensus tree from 100 trees produced by dnapars. This
would be an example of running a serial job.
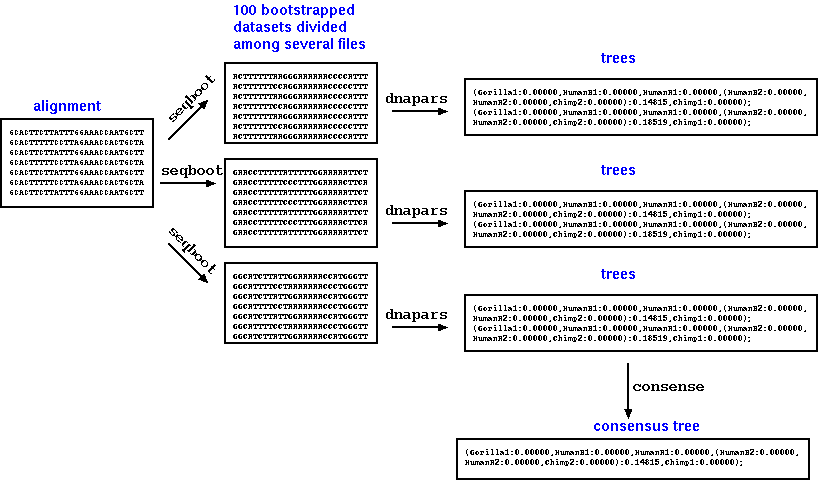
A simple script could
be written to leverage the many CPUs on the system, simply by
breaking the problem into many jobs to be run at the same time.
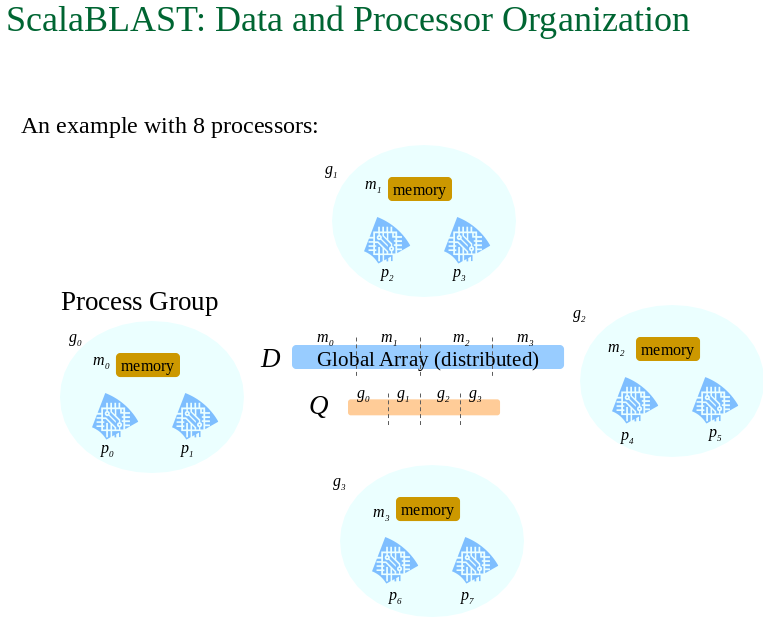
b) Parallel
processes which use multiple CPUs concurrently.
Example: ScalaBLAST
from Kalyanaraman A Introduction to BLAST
http://www.shodor.org/media/content/petascale/materials/dataIntensive/BLAST/BLAST_Intro_ppt.ppt
1. # jobs < =
number of cores - It does no good to run more jobs than
there are cores. In fact, overloading cores actually slows down
system performance, because of the added overhead of swapping
jobs on and off of the CPUs.
2. The total
size of the RAM needed for the job should not exceed
the total RAM needed by all jobs at a given time. If the
total memory used by all jobs exceeds RAM, parts of the data may
need to be swapped onto disk, and re-read at a later time. This
can drastically slow down a system.
Use the top command on
your system to get an idea of load average and memory being used
at a given time.
| load average - average number
of jobs waiting to be run on a core. When this number
exceeds the number of cores, system performance will begin
to be degraded. Memory/Swap: The system will usually try to fill the available memory. When jobs are not being run, their memory is sometimes written to a disk filesystem called Swap. Swap should be a small percentage of total memory for best performance. |
top - 13:56:29 up 64 days,
13:16, 109 users, load
average: 68.56, 67.43, 66.59 Tasks: 3015 total, 62 running, 2952 sleeping, 0 stopped, 1 zombie Cpu(s): 0.0%us, 12.9%sy, 77.4%ni, 9.5%id, 0.0%wa, 0.0%hi, 0.2%si, 0.0%st Mem: 264498644k total, 251730180k used, 12768464k free, 587932k buffers Swap: 8191996k total, 11840k used, 8180156k free, 222026532k cached PID USER PR NI VIRT RES SHR S %CPU %MEM TIME+ COMMAND 17696 umjoona7 24 4 679m 186m 26m R 101.6 0.1 80:33.99 wrf.exe 17794 umjoona7 24 4 680m 187m 26m R 101.6 0.1 93:41.31 wrf.exe 17886 umjoona7 24 4 679m 186m 26m R 101.6 0.1 94:20.41 wrf.exe 17962 umjoona7 24 4 679m 179m 24m R 101.6 0.1 94:33.43 wrf.exe 18089 umjoona7 24 4 661m 164m 26m R 101.6 0.1 94:35.67 wrf.exe 17611 umjoona7 24 4 681m 189m 27m R 101.2 0.1 92:09.04 wrf.exe 17668 umjoona7 24 4 665m 169m 28m R 101.2 0.1 89:14.75 wrf.exe 17912 umjoona7 24 4 683m 183m 24m R 101.2 0.1 94:22.77 wrf.exe 17981 umjoona7 24 4 677m 178m 24m R 101.2 0.1 94:36.04 wrf.exe 17996 umjoona7 24 4 677m 178m 24m R 101.2 0.1 94:35.98 wrf.exe 18024 umjoona7 24 4 664m 169m 27m R 101.2 0.1 94:26.51 wrf.exe 18034 umjoona7 24 4 649m 154m 26m R 101.2 0.1 94:29.06 wrf.exe 18051 umjoona7 24 4 662m 166m 26m R 101.2 0.1 94:29.71 wrf.exe 18066 umjoona7 24 4 661m 164m 26m R 101.2 0.1 94:36.35 wrf.exe |
1) CC
Unix/Linux Compute Nodes - Information Services and
Technology maintains a set of servers that may be used by any
student of staff member.
gaia, mars, mercury, venus - Login
servers for routine Linux sessions; Login by ssh or Thinlinc.
cc01, cc02, cc03...
cc12 - Linux compute nodes. These are configured
identically to venus, mars and jupiter, but should only be used
for long-running, CPU-intensive jobs. Normally, users only login
by ssh, but you can run vncserver jobs and run a full desktop
session using VNC viewer.
The U of M also operates a high capacity HPC system called Grex.
2) Digital Research Alliance of Canada (https://docs.alliancecan.ca/wiki/Technical_documentation)
| DRA is a consortium of
academic centers providing HPC services, infrastructure and
software to Canadian Researchers. Access is free of charge,
but researchers must be affiliated with a Canadian research
institution to obtain an account. |
 Unless otherwise cited or
referenced, all content on this page is licensed under
the Creative Commons License Attribution
Share-Alike 2.5 Canada Unless otherwise cited or
referenced, all content on this page is licensed under
the Creative Commons License Attribution
Share-Alike 2.5 Canada |
| PLNT4610/PLNT7690
Bioinformatics Lecture 9, part 4 of 4 |