Lecture 11, part 1 of 1
| PLNT4610/PLNT7690
Bioinformatics Lecture 11, part 1 of 1 |
The goal of genome
annotation is to identify features found in a genome, including
coding sequences for proteins and RNAs, repetitive elements and
a spectrum of other types of information found in genomes. The
end product is an annotated genome, as represented by an entry
in the GenBank database.
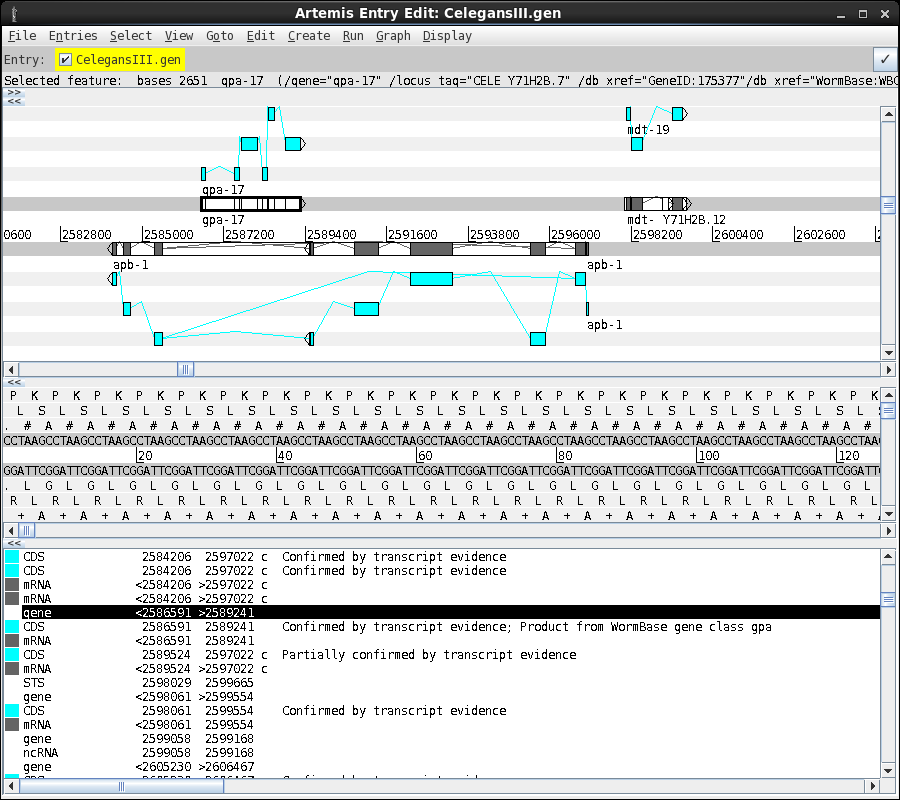
An example of an
annotated genome is shown below. A region of the C. elegans
chromosome III is shown in the artemis viewer. Note that two
genes are shown on the forward strand, and a single gene on the
reverse strand. The lower panel gives the text annotation
corresponding to features shown in the viewer.
| Genome
annotation is best described as a pipeline of steps. The example at right shows the NCBI eukaryotic genome annotation pipeline, which combines information for several sources, when available. These include the RefSeq genes and mRNA data, known protein sequences, as well as RNA seq data, if available. Coding regions are found through simple BLAST searches. The coding regions are more precisely annotated using ab initio gene prediction software to produce gene models. Gene models include precise locations of exon/intron boundaries, as inferred from mRNA or EST data. |
 from https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/ |
As with everything
else, the quality of the starting material is the single most
important factor determining the results. A low quality genome
will give low quality gene calls. Two parameters that can
indicate whether a genome is worth annotating are N50 and
percent coverage.
| N50
- N50 is the median contig size, such that 50% of the
contigs are larger than N50, and 50% are smaller. It makes
sense that if N50 contig sizes are smaller than the size
of genes, then most genes would be broken between two or
more contigs. Therefore, one guideline for a quality
assembly is to have contigs larger than the median size of
genes. Figure 1 shows a direct relationship between genome size and the median size of a gene. In terms of successful genome annotation, we need larger contigs for large eukaryotic genomes than we do for prokaryotic genomes. |
 Fig. 1 from Yandell and Ence |
Note in the above
figure that plant genes don't seem to follow this linear
relationship. Over 2 logs of genome size, gene size increases
maybe by 10%.
percent coverage
- The percentage of the genome covered by an assembly. If genome
size, as estimated from the assembly is close to the actual
genome size, then most genes will probably be found, especially
where the number of scaffolds is not large. Not getting the
entire genome may still not be a problem in eukaryotic genomes
with lots of repetitive sequences. The repetitive fraction of a
genome tends to be underestimated in incomplete genome
assemblies, because repetitive sequences often prevent the
assembly of larger contigs.
completeness of
transcriptome - Not all mRNAs are expressed in all
tissues. Since transcripts from the transcriptome are part of
the annotation process, many genes could be missed if they are
not present in one or more of the tissues, developmental stages
or conditions used in assembly of the transcriptome.
Because much of the annotation process relies upon similarity comparisons, it is critical to mask repeats within your genomic sequences. Two types of repeats need to be masked:
|
Original
sequence Soft
masking Hard
masking |
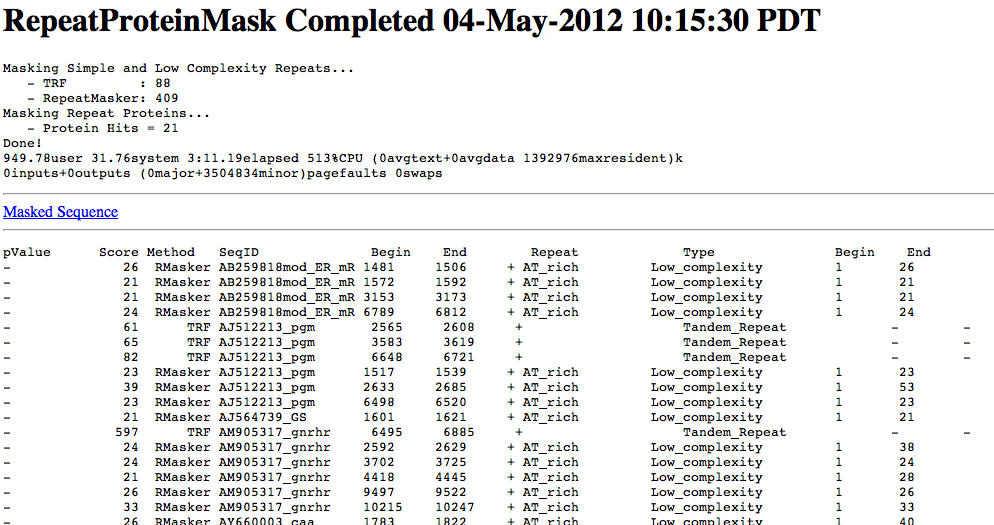
This step amounts to the discovery of protein coding regions, often annotated as CDS regions.
| Similarity
search programs such as BLASTX can compare a genomic
nucleotide sequence with a protein database to find
potential coding regions. In the example at right, output for a 9.5 kb contig from a fungal genome shows three potential coding regions. The right most coding region appears to include an intron between two CDS regions. |
 |
Similarity alignment results only give the approximate locations of CDS features. The precise exon/intron structure of a gene, along with other features such as ribosome binding sites, 5'UTRs, 3'UTRs or promoter regions, require more sophisticated gene prediction tools. The goal is to create a model of a gene describing these features with precise begin and end points.
For example, splice
junctions are predicted using tools such as splign. Splign
predicts exon/intron structure based on cDNA/EST sequences found
in genome databases. Splign works in three steps:
| In
the example, the sequence of an exon boxed in green is
shown. The cDNA is on top (pink) and the chromosomal
sequence is on the bottom (blue). The AG of the splice acceptor 5' to exon 5, and the GT of the donor 3' to exon 5 are highlighted in green. |

from https://www.ncbi.nlm.nih.gov/Web/Newsltr/V14N2/splign.html
|
| Annotation
pipelines such as MAKER
combine information from many sources, including gene
predictors, BLAST, and repeat maskers, to create
integrated alignments. At right is an gene model, viewed
in the Apollo
genome viewer. |
 from http://gmod.org/wiki/MAKER |
| The
results of genome annotation are usually saved in GFF3
files. GFF3 is an extensive format that can represent
locations strands and identifiers for just about any
feature that can be annotated. It is a generally universal
feature format, but there are issues, with specific
software applications, which may not be fully compliant
with GFF3. As well, there are some conflicts between NCBI
annotation and GFF3*. The specifications for GFF3 can be found at http://gmod.org/wiki/GFF3 |
0 ##gff-version 3 1 ##sequence-region ctg123 1 1497228 2 ctg123 . gene 1000 9000 . + . ID=gene00001;Name=EDEN 3 ctg123 . TF_binding_site 1000 1012 . + . ID=tfbs00001;Parent=gene00001 4 ctg123 . mRNA 1050 9000 . + . ID=mRNA00001;Parent=gene00001;Name=EDEN.1 5 ctg123 . five_prime_UTR 1050 1200 . + . Parent=mRNA0001 6 ctg123 . CDS 1201 1500 . + 0 Parent=mRNA0001 7 ctg123 . CDS 3000 3902 . + 0 Parent=mRNA0001 8 ctg123 . CDS 5000 5500 . + 0 Parent=mRNA0001 9 ctg123 . CDS 7000 7600 . + 0 Parent=mRNA0001 10 ctg123 . three_prime_UTR 7601 9000 . + . Parent=mRNA0001 11 ctg123 . mRNA 1050 9000 . + . ID=mRNA00002;Parent=gene00001;Name=EDEN.2 12 ctg123 . five_prime_UTR 1050 1200 . + . Parent=mRNA0002 13 ctg123 . CDS 1201 1500 . + 0 Parent=mRNA0002 14 ctg123 . CDS 5000 5500 . + 0 Parent=mRNA0002 15 ctg123 . CDS 7000 7600 . + 0 Parent=mRNA0002 16 ctg123 . three_prime_UTR 7601 9000 . + . Parent=mRNA0002 17 ctg123 . mRNA 1300 9000 . + . ID=mRNA00003;Parent=gene00001;Name=EDEN.3 18 ctg123 . five_prime_UTR 1300 1500 . + . Parent=mRNA0003 19 ctg123 . five_prime_UTR 3000 3300 . + . Parent=mRNA0003 20 ctg123 . CDS 3301 3902 . + 0 Parent=mRNA0003 21 ctg123 . CDS 5000 5500 . + 2 Parent=mRNA0003 22 ctg123 . CDS 7000 7600 . + 2 Parent=mRNA0003 23 ctg123 . three_prime_UTR 7601 9000 . + . Parent=mRNA0003 |
 Unless otherwise cited or
referenced, all content on this page is licensed under
the Creative Commons License Attribution
Share-Alike 2.5 Canada Unless otherwise cited or
referenced, all content on this page is licensed under
the Creative Commons License Attribution
Share-Alike 2.5 Canada |
| PLNT4610/PLNT7690
Bioinformatics Lecture 11, part 1 of 1 |