WebLogo 3: User's Manual
Contents
Release Notes and Known Bugs
The WebLogo release notes detail changes to WebLogo and known issues with particular versions.Introduction
WebLogo is a web based application designed to make the generation of sequence logos as easy and painless as possible.
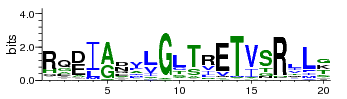
A sequence logo is a graphical representation of an amino acid or nucleic acid multiple sequence alignment. Each logo consists of stacks of symbols, one stack for each position in the sequence. The overall height of the stack indicates the sequence conservation at that position, while the height of symbols within the stack indicates the relative frequency of each amino or nucleic acid at that position. The width of the stack is proportional to the fraction of valid symbols in that position. (Positions with many gaps have thin stacks.)In general, a sequence logo provides a richer and more precise description of, for example,a binding site, than would a consensus sequence.
References
Crooks GE, Hon G, Chandonia JM, Brenner SE WebLogo: A sequence logo generator, Genome Research, 14:1188-1190, (2004) [ Full Text ]
Schneider TD, Stephens RM. 1990. Sequence Logos: A New Way to Display Consensus Sequences. Nucleic Acids Res. 18:6097-6100
Creating Sequences Logos using the Web interface
- Sequence Data
- Enter your multiple sequence alignment or position weight matrix file, or select a file to upload. Supported file formats include CLUSTALW, FASTA, plain flatfile, MSF, NBRF, PIR, NEXUS and PHYLIP for multiple sequence alignments, and transfac for position weight matrices. All sequences must be the same length, else WebLogo will return an error and report the first sequence that differed in length from previous sequences.
- Output format
- Generally speaking, vector formats (EPS and PDF) are better for print, while bitmaps (JPEG and PNG) are more suitable for displaying on the screen or embedding into a web page.
-
PNG: (600 DPI) Print resolution bitmap PNG: (low res, 96 DPI) Screen resolution bitmap JPEG: Screen resolution bitmap EPS: Encapsulated postscript PDF: Portable Document Format SVG: Scalable Vector Graphics - Logo size
- The physical dimensions of the generated logo. Specifically, Logo Size controls the size of individual symbols stacks.
-
small: 5.4 points wide (same as 9pt Courier), aspect ratio 5:1 medium: Double the width and height of small. large: Triple the width and height of small. - The choices have been limited to promote inter-logo consistency. Small logos can fit 80 stacks across a printed page, or 40 across a half page column. The command line interface provides greater control, if so desired.
- Stacks per line
- If the length of the sequences is greater than this maximum number of stacks per line, then the logo will be split across multiple lines.
- Sequence Type
- Selects the type of biological molecule.
auto: Automatically guess sequence type from the data protein dna rna - Ignore Lower Case
- Disregard lower case letters and only count upper case letters in sequences
- Units
- The units used for the y-axis.
-
probability: Show residue probabilities, rather than information content. If compositional adjustment is disabled, then these are the raw residue frequencies. bits: Information content in bits nats: Natural units, 1 bit = ln 2 (0.69) nats kT: Thermal energy units in natural units (numerically the same as nats) kJ/mol: Thermal energy (assuming T = 300 K) kcal/mol: Thermal energy (assuming T = 300 K) - First position number
- The numerical label of the first position in the sequence data in the input file. The label must be an integer. Residue labels for the logo will be relative to this number. (See also: Logo range.)
- Logo range
- By default, all sequence data from the input file is displayed in the Sequence Logo. With this option, you can instead show a subrange of the sequence data. The numbering of Start and End Positions is relative to the First Position Number. Thus, if the First Position Number is "2", Start is "5" and End is "10", then the 4th through 9th (inclusive) sequence positions of the input file will be displayed, and they will be numbered "5", "6", "7", "8", "9" and "10".
- Composition
-
The background composition of the genome or proteome from which the sequences have been drawn. The default, automatic option is to use equiprobable background for nucleic acids and a typical amino acid usage pattern for proteins. However, you may also explicitly set the expected CG content for nucleic acid sequences, insist on equiprobable background distributions, or turn off composition adjustment altogether.
Compositional adjustment has two effects. First, the information content of a site is defined as the relative entropy of the monomers at that site to the background distribution. Consequentially, rare monomers have higher information content (when they occur) than relatively common monomers.
Secondly, the background composition is used in the small sample correction of information content. Briefly, if only a few sequences are available in the multiple sequence alignment, then sites typically appear more conserved than they really are. Small samples bias the relative entropy upwards. To compensate, we add pseudocounts to the actual counts, proportional to the expected background composition. These pseudocounts smooth the data for small samples, but become irrelevant for large samples. The proportionality constant is set to 4 for nucleic acid sequences, and 20 for proteins (these numbers have been found to give reasonable results in practice).
Behind the scenes, things are more complex. We do a full Bayesian calculation, starting with explicit Dirichlet priors based on the background composition, to which we add the data and then calculate both the posterior mean relative entropy (the stack height) and Bayesian 95% confidence intervals for error bars. These interesting details will be explained elsewhere.
- Scale stack width
- Scale the visible stack width by the fraction of symbols in the column.(I.e. columns with many gaps or unknown residues are narrow.)
- Error bars
- Display error bars. These indicate an approximate Bayesian 95% confidence interval.
- Title
- Give your logo a title.
- Figure label
- An optional figure label, added to the top left (e.g. '(a)').
- X-axis
- Add a label to the x-axis, or hide axis altogether.
- Y-axis
- The vertical axis indicates the information content of a sequence position. Use this option to toggle the y-axis and override the default axis label.
- Y-axis scale
- The height of the y-axis in designated units. The automatic option will pick reasonable defaults based on the sequence type and axis unit.
- Y-axis tic spacing
- The distance between major tic marks on the y-axis.
- Sequence end labels
- Choose this option to label the 5' & 3' ends of nucleic acid or the N & C termini of amino acid sequences.
- Version fineprint
- Toggle display of the WebLogo version information in the lower right corner. Optional, but we appreciate the acknowledgment.
- Auto:
- Use Base Pairing for nucleic acids (NA), Hydrophobicity for amino acids (AA).
- Monochrome:
- All symbols black
- Base Pairing
(NA default): -
2 Watson-Crick hydrogen bonds TAU dark orange 3 Watson-Crick hydrogen bonds GC blue - Classic (NA):
-
WebLogo (version 1 and 2) and makelogo default color scheme for nucleic acids: G, orange; T & U, red; C, blue; and A, green.G orange TU red C blue A green - Hydrophobicity
(AA default): -
Hydrophilic RKDENQ blue Neutral SGHTAP green Hydrophobic YVMCLFIW black - Chemistry (AA):
- Color amino acids according to chemical properties.
-
WebLogo (version 1 and 2) and makelogo default color. (Note that the WebLogo 2 documentation erroneously lists Q and N under green.)Polar G,S,T,Y,C green Neutral Q,N purple Basic K,R,H blue Acidic D,E red Hydrophobic A,V,L,I,P,W,F,M black - Charge (AA) :
-
Positive KRH blue Negative DE red - Custom:
- A custom color scheme can be specified in the input field below. Specify colors on the left and associated symbols on the right. Colors are entered using CSS2 (Cascading Style Sheet) syntax. (E.g. 'red', '#F00', '#FF0000', 'rgb(255, 0, 0)', 'rgb(100%, 0%, 0%)' or 'hsl(0, 100%, 50%)' for the color red.)
- More Options
-
The weblogo command line client,
weblogo, provides many more options and greater control over the final logo appearance.
Installing WebLogo
-
Dependencies
- WebLogo version 3 is written in python. It is necessary to have Python 3.7 or later and the extension package numpy installed before WebLogo will run. WebLogo also requires a recent version of ghostscript to create PNG and PDF output, and pdf2svg to generate SVG output.
-
Download and Installation
-
WebLogo and its dependencies can be downloaded and installed with pip:
pip install weblogoAlternatively, weblogo and its dependencies can be installed manually. The WebLogo source code can be downloaded from Weblogo's Github repository. This code is distributed under various open source licenses. Please consult theLICENSE.txtfile in the source distribution for details.After unpacking the WebLogo tarfile, it should be possible to immediately create logos using the command line client (provided that python, numpy and ghostscript have already been installed).
./weblogo --format PNG < cap.fa > cap.pngPlease consult the file
build_examples.shfor more examples.To run WebLogo as a stand alone web service, run the logo server command :
./weblogo --serveIt should now be possible to access WebLogo at http://localhost:8080/.
-
Web App
-
To use WebLogo as a web application, first install the weblogo dependencies and libraries as above, then place (or link) the
weblogo/htdocsdirectory somewhere within the document root of your webserver. The webserver must be able to execute the CGI scriptcreate.cgi. For Apache, you may have to add anExecCGIoption and add a cgi handler in thehttpd.confconfiguration file. Something like this:DocumentRoot "/home/ec2-user/weblogo/weblogo/htdocs" # Further relax access to the default document root: <Directory "/home/ec2-user/weblogo/weblogo/htdocs"> Options Indexes FollowSymLinks ExecCGI MultiViews AddHandler cgi-script .cgi AllowOverride All Require all granted </Directory>It may also be necessary to set thePATHandPYTHONPATHenvironment variables.SetEnv PYTHONPATH /path/to/weblogo/librariesThe cgi script also has to be able to find the 'gs' ghostscript executable. The maximum bytes of uploaded sequence data can be controlled with theWEBLOGO_MAX_FILE_SIZEenvironment variable.SetEnv WEBLOGO_MAX_FILE_SIZE 1000000
-
weblogo, The WebLogo Command Line Interface (CLI) -
The command line client has many options not available through the web interface. Please consult the bundled
build_examples.shscript for inspiration.Usage: weblogo [options] < sequence_data.fa > sequence_logo.eps Create sequence logos from biological sequence alignments. Options: --version show program's version number and exit -h --help show this help message and exit Input/Output Options: -f --fin FILENAME Sequence input file (default: stdin) --upload URL Upload input file from URL -D --datatype FORMAT Type of multiple sequence alignment or position weight matrix file: (clustal, fasta, msf, genbank, nbrf, nexus, phylip, stockholm, intelligenetics, table, array, transfac) -o --fout FILENAME Output file (default: stdout) -F --format FORMAT Format of output: eps (default), png, png_print, pdf, jpeg, svg, logodata Logo Data Options: -A --sequence-type TYPE The type of sequence data: 'protein', 'rna' or 'dna'. -a --alphabet ALPHABET The set of symbols to count, e.g. 'AGTC'. All characters not in the alphabet are ignored. If neither the alphabet nor sequence-type are specified then weblogo will examine the input data and make an educated guess. See also --sequence-type, --ignore-lower-case -U --units UNIT A unit of entropy ('bits' (default), 'nats', 'digits'), or a unit of free energy ('kT', 'kJ/mol', 'kcal/mol'), or 'probability' for probabilities --composition COMP. The expected composition of the sequences: 'auto' (default), 'equiprobable', 'none' (do not perform any compositional adjustment), a CG percentage, a species name (e.g. 'E. coli', 'H. sapiens'), or an explicit distribution (e.g. "{'A':10, 'C':40, 'G':40, 'T':10}"). The automatic option uses a typical distribution for proteins and equiprobable distribution for everything else. --weight NUMBER The weight of prior data. Default depends on alphabet length -i --first-index INDEX Index of first position in sequence data (default: 1) -l --lower INDEX Lower bound of sequence to display -u --upper INDEX Upper bound of sequence to display Transformations: Optional transformations of the sequence data. --ignore-lower-case Disregard lower case letters and only count upper case letters in sequences. --reverse reverse sequences --complement complement nucleic sequences --revcomp reverse complement nucleic sequences Logo Format Options: These options control the format and display of the logo. -s --size LOGOSIZE Specify a standard logo size (small, medium (default), large) -n --stacks-per-line COUNT Maximum number of logo stacks per logo line. (default: 40) -t --title TEXT Logo title text. --label TEXT A figure label, e.g. '2a' -X --show-xaxis YES/NO Display sequence numbers along x-axis? (default: True) -x --xlabel TEXT X-axis label --annotate TEXT A comma separated list of custom stack annotations, e.g. '1,3,4,5,6,7'. Annotation list must be same length as sequences. --rotate-numbers YES/NO Draw X-axis numbers with vertical orientation (default: False). --number-interval NUMBER Distance between numbers on X-axis (default: 5) -S --yaxis NUMBER Height of yaxis in units. (Default: Maximum value with uninformative prior.) -Y --show-yaxis YES/NO Display entropy scale along y-axis? (default: True) -y --ylabel TEXT Y-axis label (default depends on plot type and units) -E --show-ends YES/NO Label the ends of the sequence? (default: False) -P --fineprint TEXT The fine print (default: weblogo version) --ticmarks NUMBER Distance between ticmarks (default: 1.0) --errorbars YES/NO Display error bars? (default: True) --reverse-stacks YES/NO Draw stacks with largest letters on top? (default: True) Color Options: Colors can be specified using CSS2 syntax. e.g. 'red', '#FF0000', etc. -c --color-scheme SCHEME Specify a standard color scheme (auto, base pairing, charge, chemistry, classic, hydrophobicity, monochrome) -C --color COLOR SYMBOLS DESCRIPTION Specify symbol colors, e.g. --color black AG 'Purine' --color red TC 'Pyrimidine' --default-color COLOR Symbol color if not otherwise specified. Font Format Options: These options provide control over the font sizes and types. --fontsize POINTS Regular text font size in points (default: 10) --title-fontsize POINTS Title text font size in points (default: 12) --small-fontsize POINTS Small text font size in points (default: 6) --number-fontsize POINTS Axis numbers font size in points (default: 8) --text-font FONT Specify font for labels (default: ArialMT) --logo-font FONT Specify font for logo (default: Arial-BoldMT) --title-font FONT Specify font for title (default: ArialMT) Advanced Format Options: These options provide fine control over the display of the logo. -W --stack-width POINTS Width of a logo stack (default: 10.8) --aspect-ratio POINTS Ratio of stack height to width (default: 5) --box YES/NO Draw boxes around symbols? (default: no) --resolution DPI Bitmap resolution in dots per inch (DPI). (Default: 96 DPI, except png_print, 600 DPI) Low resolution bitmaps (DPI<300) are antialiased. --scale-width YES/NO Scale the visible stack width by the fraction of symbols in the column? (I.e. columns with many gaps of unknowns are narrow.) (Default: yes) --debug YES/NO Output additional diagnostic information. (Default: False) --errorbar-fraction NUMBER Sets error bars display proportion (default: 0.9) --errorbar-width-fraction NUMBER Sets error bars width display proportion (default: 0.25) --errorbar-gray NUMBER Sets error bars' gray scale percentage (default: 0.75) WebLogo Server: Run a standalone webserver on a local port. --serve Start a standalone WebLogo server for creating sequence logos. --port PORT Listen to this local port. (Default: 8080)
WebLogo Application Programmer Interface (API)
The WebLogo python libraries provide even greater flexibility than the command line client. Please consult the code documentation at readthedocs.WebLogo Development and Future Features
The development project is hosted at https://github.com/WebLogo/weblogo. If you wish to extend WebLogo or to contribute code, then you should download the full source code development package directly from the Github repository.
> git clone https://github.com/WebLogo/weblogo
> pip install -e .
> cd weblogo
Please consult the developer notes, DEVELOPERS.txt and software license LICENSE.txt
Outstanding bugs and feature requests are listed on the WebLogo issue tracker.
Miscellanea
-
WebLogo 2
- The legacy WebLogo 2 sever can be found here.
-
Acknowledgments
-
WebLogo was created by Gavin E. Crooks, Liana Lareau, Gary Hon, John-Marc Chandonia and Steven E. Brenner. Many others have provided suggestions, bug fixes and moral support.
-
WebLogo was originally based upon the programs alpro and makelogo, both of which are part of Tom Schneider's delila package. Many thanks are due to him for making this software freely available and for encouraging its use.
-
Disclaimer
-
While no permanent records are kept of submitted sequences, we cannot undertake to guarantee that data sent to WebLogo remains secure. Moreover, no guarantees whatsoever are provided about data generated by WebLogo.
-
Feedback
-
Suggestions on how to improve WebLogo are heartily welcomed! Please direct questions to WebLogo's issue tracker.