TUTORIAL: MULTIPLE ALIGNMENTS |
Nov. 18, 2024 |
TUTORIAL: MULTIPLE ALIGNMENTS |
Nov. 18, 2024 |
| Warning! Most multiple alignment programs will align either DNA or amino acid sequences. However, it's important to know that unless nucleic acid sequences are very closely-related, with few gaps (eg. tRNA, rRNA genes), a reliable multiple alignment (or for that matter, even a pairwise alignment) is almost impossible. The reason is that nucleic acids use a 4-letter alphabet, allowing many equally good alignments to form a set of sequences. In contrast, the 20-letter amino acid alphabet drastically decreases the number of possible alignments, so that an obvious 'correct' alignment is usually possible to find. |
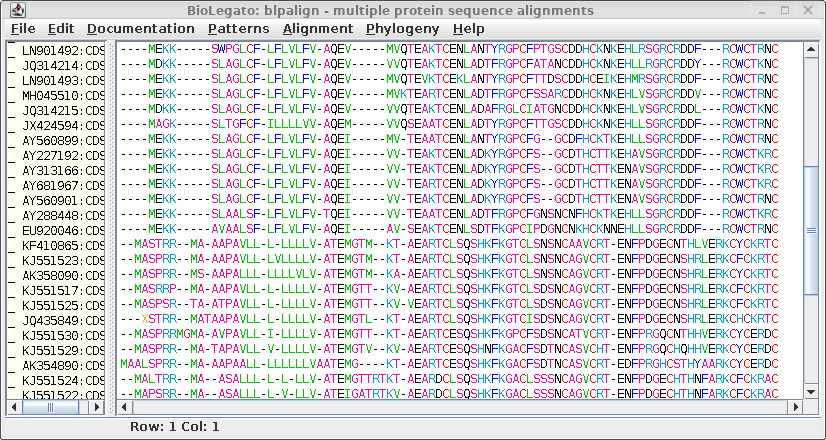
| Select all amino acid sequences and open the Alignment menu, which shows programs related to multiple sequence alignment | 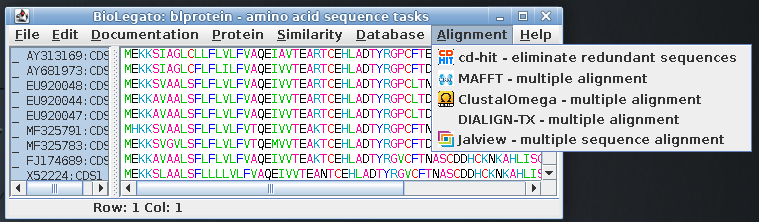 |
| # comparing sequences
from
0
to 81 ---------- new table with 60 representatives 81 finished 60 clusters |
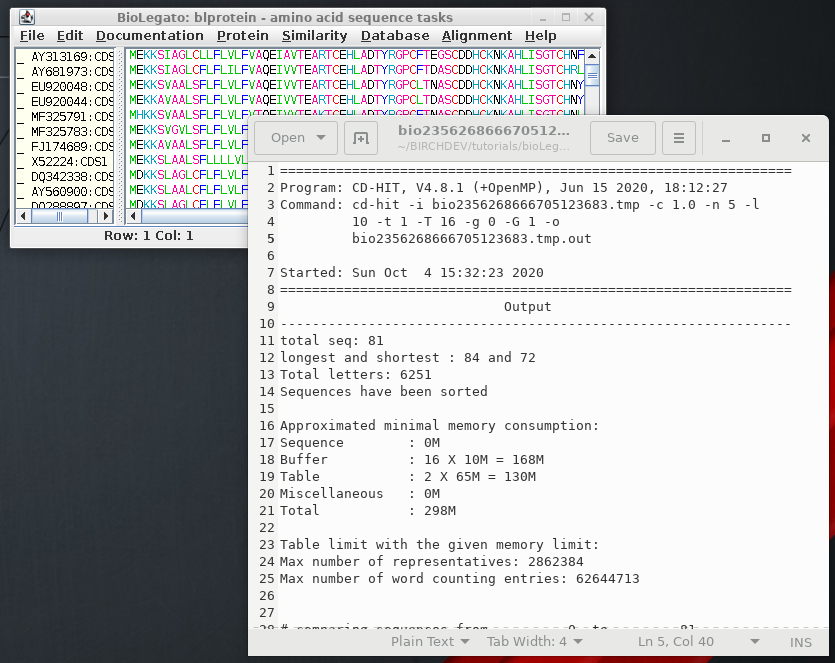
This indicates
that of 81 sequences in the dataset, 60 clusters were found
with 1 or more members. Using an identity threshold of 1.0,
all sequences in a cluster must be 100% identical |
| >Cluster 21 0 80aa, >AP006168:CDS20... * 1 80aa, >MF580139:CDS1... at 100.00% >Cluster 22 0 80aa, >HG792392:CDS1... * 1 80aa, >JF797205:CDS1... at 100.00% 2 80aa, >EF200066:CDS1... at 100.00% >Cluster 23 0 80aa, >EU952901:CDS1... * >Cluster 24 0 80aa, >EU958628:CDS1... * 1 80aa, >EU957417:CDS1... at 100.00% 2 80aa, >EU958657:CDS1... at 100.00% 3 80aa, >EU961911:CDS1... at 100.00% |
Output for four
of the clusters are shown. For example, cluster 21 contains
two sequences. AP006168:CDS20 was retained, and
MF580139:CDS1 was deleted. Similarly for Cluster 22, HG792392:CDS1 was retained while the other two were deleted. |
| Select the non-redundant sequences resulting from CD-HIT, and choose Alignment --> Clustal Omega. | 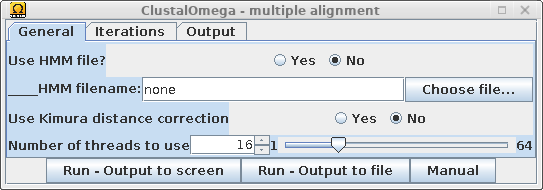 |
| Clustal sends the alignment to a new blpalign
window. Choose File --> Save ALL As and save the protein alignment as defensin.pro.cdhit.clustalo.fsa |
 |
 Warning!: Do not use guide
trees generated by clustal or other multiple alignment
programs for any purpose eg. phylogenetic analysis of
sequence or species evolution. These trees are based
on pairwise alignments, and therefore do not contain the
evolutionary information found in the gaps that are present
in the completed alignment. Once you have an alignment, you
can then go back and construct a phylogenetic tree. Warning!: Do not use guide
trees generated by clustal or other multiple alignment
programs for any purpose eg. phylogenetic analysis of
sequence or species evolution. These trees are based
on pairwise alignments, and therefore do not contain the
evolutionary information found in the gaps that are present
in the completed alignment. Once you have an alignment, you
can then go back and construct a phylogenetic tree. |
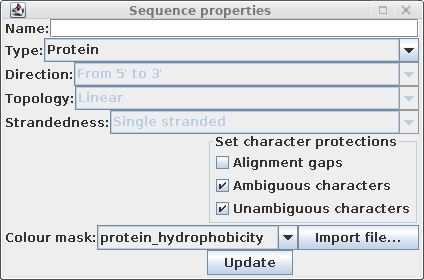
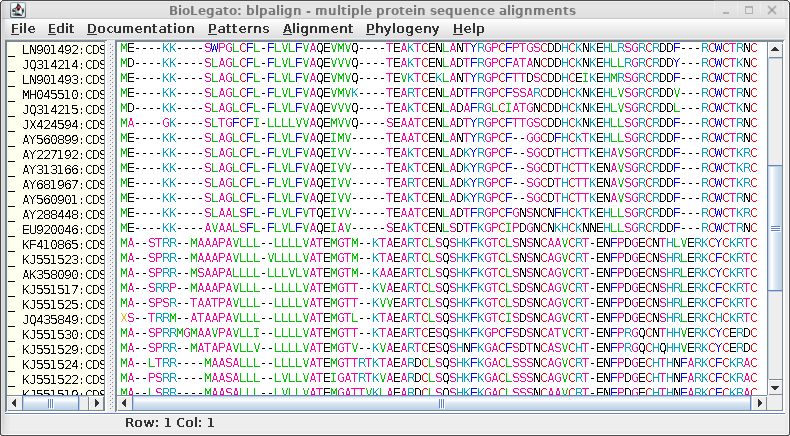
| Choose Edit --> Select All, and then Edit
--> Get Info. Change the Colour mast to
protein_hydrophobicity. (Note: If the colors don't change, either resize the BioLegato window or scroll a bit using one of the scroll bars. This forces BioLegato to redraw the sequence canvas.) |
 |
MAFFT is a
program that implements a wide variety of alignment strategies.
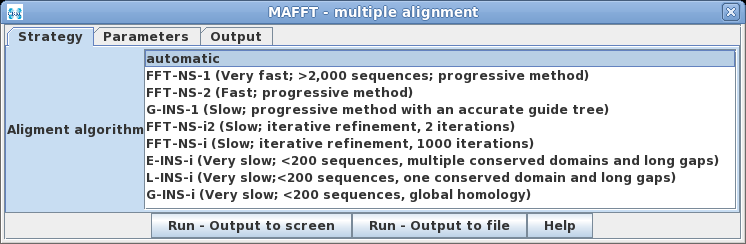
| One of the unique things to note about MAFFT
is that it will automatically choose the best algorithm for
alignment, based on the number of input sequences, unless a
method is specified. The methods cover most of the
situations typically encountered in multiple alignments. |
 |
Because
the type of problem will vary is is best to consult the MAFFT
Algorithms page to see which algorithm best applies to
your specific dataset. For relatively small numbers of sequences
(eg. < 200 sequence) methods such as E-INS-i, L-INS-i and
GINS-i construct a guide tree based on pairwise distances and
then traverse the tree until all sequences have been added,
similar to Clustal Omega.The faster FFT--NS-1 and FFT-NS-2
estimate pairwise sequence distances based on frequencies of
6-mer oligonucleotides or oligopeptides shared between each pair
of sequences. Methods also vary depending on whether or not they
refine the initial alignment by iteratively aligning subsets of
aligned sequences, and by whether they recalculate the guide
tree based on the first alignment, and then repeat the
alignment. Obviously, for short numbers of sequences, the slower
more accurate methods are preferred.
To launch
MAFFT, choose Alignment --> MAFFT. Output is
sent directly to blpalign. Try comparing the output using the
FFT-NS-2 and FFT-NS-i, which essentially compares a fast
progressive method with a slower iterative method.
| Weblogo has many options for presentation of
the logo. For now, we'll just add a title to the Logo title box. Note that one quirk of Weblogo is that it doesn't allow blank spaces in titles. To separate words, simply use an underscore (_) wherever a blank would normally be used. The title here is "Plant_Defensins" |
 |
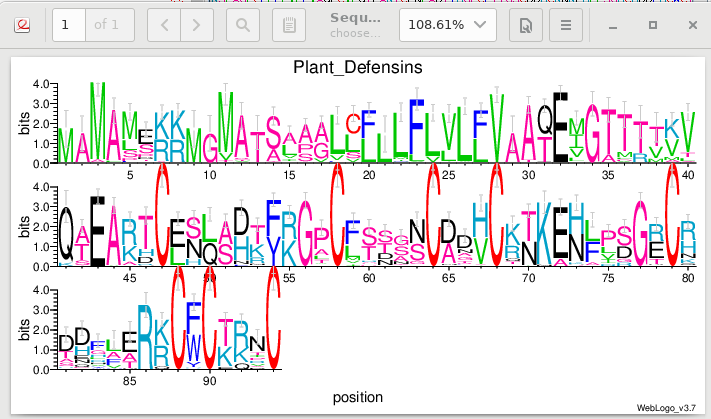
| Weblogo plots information content of each
position in the alignment in bits of information. The
information content can be thought of as a deviation from
randomness. A high bit score would be a highly unlikely
outcome, typically occurring where specific amino acids are
conserved. A low bit score would mean closer to a random
distribution of amino acids at a position, typically at less
conserved positions. Note that we can see fairly high bit scores at the K/R positions mentioned earlier. The heights of the letters indicate the relative frequencies of each amino acid, at that position in the alignment. |
 |
pal2nal.pl by Mikita Suyama aligns DNA sequences using the
corresponding amino acid alignment as a guide. Thus, if you have
aligned a set of amino acid sequences, it is straightforward to
generate the corresponding DNA alignment. pal2nal.pl requires two
files for input, in FASTA format: a file containing the unaligned
DNA coding sequences, and the second file containing the
corresponding amino acid sequences, aligned by a program such as
Clustal Omega or MAFFT. When we run pal2nal.pl from blpalign,
blpalign will generate a DNA alignment file from the proteins
selected. Thus, all the user needs to do is to select a file
containing the corresponding DNA CDS sequences, unaligned. The
following example illustrates this process.
| Notes: 1) pal2nal.pl needs the DNA and aligned amino acid sequences to have the same names. During the extraction process, during translation, the names may be modified, so it may be necessary to change names in 'File --> Get Info', before you export to a .fsa file 2) Where two or more copies of a gene are present in a single entry (eg. CAGTHIOGN:CDS1 and CAGTHIOGN:CDS2), it is necessary to give them each unique names so that pal2nal.pl can distinguish them. Since the CDS extensions will be removed when pal2nal.pl is run, one solution is to delete CDS but retain the number (eg CAGTHIOGN_1 and CAGTHIOGN_2). The current BioLegato instances implementation of Clustal Omega and MAAFT usually handle these steps automatically. Nonetheless, if you are having problems with pal2nal.pl, make sure that the names of sequences are the same for both protein and nucleic acid sequences. 3) Before running pal2nal.pl, blpalign compares the list of sequence names in the protein alignment with the names in the DNA file. Only DNA sequences with a corresponding protein in the alignment will be used as input for pal2nal.pl. This is useful, because it means that different subsets of the aligned sequences could be aligned by pal2nal.pl, using a single unaligned DNA file. |
Using the MAAFT FFT-NS-i alignment as an example, choose Edit
--> Select All.
| To run pal2nal.pl, choose Alignment --> pal2nal,
and choose the DNA filename. |
 |
The aligned DNA sequences appear in a new blnalign window:
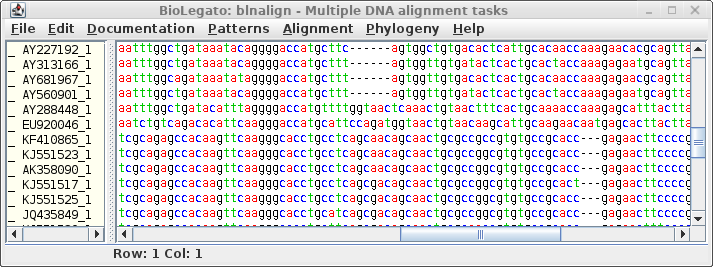
Again we see the two subfamilies of defensins, with codons aligned
to the corresponding amino acids in the protein alignment. This
alignment could be used directly for phylogenetic analysis.
Alignments can be edited directly in blpalign or blnalign. By
default, only gaps can be edited out, although it is possible to
delete amino acids or nucleotides if protections are changed (Edit --> GetInfo).
Shifting gaps for a group of sequences in an alignment could
potentially improve the alignment.
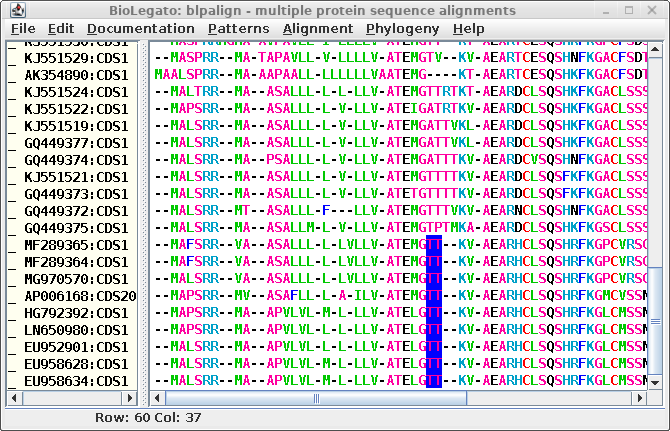
| The T's highlighted in blue could just as
legitimately line up with T's in the sequences above. In
other words, it might be just as good an alignment if the
GTT-- motif was changed to G--TT. As long as the number of
gaps is conserved, the alignment could easily be revised. |
 |
|
We can move the block over as follows. First select all of the sequences in the block by name. Click on the topmost sequence in the block, MF289365, hold the shift key, and click on EU958634. We can make these sequences function as a group by choosing Edit --> Group. The blpalign window will now look like this: |
 |
| First, insert two gap
characters (--) in any sequence in group 1 to shift
these amino acids to line up TT with the other occurrences
of TT. You will notice that insertion of the gaps puts the rest of the sequences downstream of the insertion out of alignment. |
 |
| To restore the alignment, use the Backspace
or Delete key to remove the two gaps to the right of the
TTs. You can now see the amino acid motifs KV-AEA... lining up with the rest of the sequences. |
 |
In many cases you need an alignment displayed as editable text.
This might be true if you wanted to be able to import the
alignment into a word-processor or HTML editor for further
modification, such as coloring or underlining certain
characters.
| As an example, we'll print out just the
sequences in group 1 from the previous section. Choose Alignment --> REFORM. |
 |
By default, the output will print a consensus sequence at top,
and only show in the alignment those amino acids that differ from
the consensus.
Various features of the output can be changed in the REFORM menu For example, to print ALL amino acids at every position, set Print conserved sites in alignment as dots to No.
10 20 30 40 50 60 70
--MAxSRR--ma--ApvlvL-l-l-lLV-ATElG--TTKV-AEARHCLSQSHRFKGlCmsSnNCANvCqT
MF289365:C....f.....v....sa.l.....v.......m.......................p.vr.g......k.
MF289364:C....f.....v....sa.l.....v.......m.......................p.vr.g......k.
MG970570:C....l.....v....sa.l.....v.......m.......................p.vr.g......k.
AP006168:C....p......v...safl....a.i......m.......................m.v.........r.
HG792392:C....p................m................................................
LN650980:C....p.............................................................a...
EU952901:C....l.................................................................
EU958628:C....l................m................................................
EU958634:C....l................m................................................
80 90
-EnFPgGECkaeGatRKCFCKkiC
MF289365:C.....d...qtq.le......rv.
MF289364:C.....d...qtq.lq......rv.
MG970570:C.....d...qtq.le......rv.
AP006168:C..s..d....sh.le.......v.
HG792392:C........................
LN650980:C........................
EU952901:C........................
EU958628:C........................
EU958634:C.........r..............
b) JALVIEW - Graphic display and alignment
10 20 30 40 50 60 70
--MAxSRR--ma--ApvlvL-l-l-lLV-ATElG--TTKV-AEARHCLSQSHRFKGlCmsSnNCANvCqT
MF289365:C--mafsrr--va--asalll-l-lvllv-atemg--ttkv-aearhclsqshrfkgpcvrsgncanvckt
MF289364:C--mafsrr--va--asalll-l-lvllv-atemg--ttkv-aearhclsqshrfkgpcvrsgncanvckt
MG970570:C--malsrr--va--asalll-l-lvllv-atemg--ttkv-aearhclsqshrfkgpcvrsgncanvckt
AP006168:C--mapsrr--mv--asafll-l-a-ilv-atemg--ttkv-aearhclsqshrfkgmcvssnncanvcrt
HG792392:C--mapsrr--ma--apvlvl-m-l-llv-atelg--ttkv-aearhclsqshrfkglcmssnncanvcqt
LN650980:C--mapsrr--ma--apvlvl-l-l-llv-atelg--ttkv-aearhclsqshrfkglcmssnncanacqt
EU952901:C--malsrr--ma--apvlvl-l-l-llv-atelg--ttkv-aearhclsqshrfkglcmssnncanvcqt
EU958628:C--malsrr--ma--apvlvl-m-l-llv-atelg--ttkv-aearhclsqshrfkglcmssnncanvcqt
EU958634:C--malsrr--ma--apvlvl-m-l-llv-atelg--ttkv-aearhclsqshrfkglcmssnncanvcqt
80 90
-EnFPgGECkaeGatRKCFCKkiC
MF289365:C-enfpdgecqtqglerkcfckrvc
MF289364:C-enfpdgecqtqglqrkcfckrvc
MG970570:C-enfpdgecqtqglerkcfckrvc
AP006168:C-esfpdgeckshglerkcfckkvc
HG792392:C-enfpggeckaegatrkcfckkic
LN650980:C-enfpggeckaegatrkcfckkic
EU952901:C-enfpggeckaegatrkcfckkic
EU958628:C-enfpggeckaegatrkcfckkic
EU958634:C-enfpggecraegatrkcfckkic
Jalview is a feature-rich sequence alignment analysis system. It
can be launched by selecting an alignment in blnalign or blpalign,
and choosing 'Alignment --> Jalview'. (Note: since
Jalview also performs multiple alignments, bldna and blprotein
also have options to launch Jalview).
The alignment above is shown using one of several color schemes
available. The Blosum62 color scheme shows conservation, as
measured by Blosum62 matrix scores at each position.
The alignment can be written in paginated form, suitable for
printing, by saving in a variety of formats.
Jalview can also do complete alignments from unaligned sequences.
For a full description of the capabilities of Jalview, see http://www.jalview.org/help.html.