TUTORIAL: Simulated Cloning |
Oct. 21, 2014 |
TUTORIAL: Simulated Cloning |
Oct. 21, 2014 |
Any recombinant construct can be simulated by pasting together
the correct sequences into a single file.
One easy way is to use NUMSEQ to print out the precise fragments
required, and paste them, in the correct order, into a file, using
any text editor.
EXAMPLE: Cloning beta-glucuronidase gene (GUS) from pBI101 to pBluescriptKSm13+.
The GUS gene in pBI101 can be conveniently excised using BamHI
and SacI (see map).
The goal is to make a datafile that correctly represents the
recombinant construct that results from cloning the BamHI/SacI
fragment containing the GUS gene into the BamHI/SacI-digested
BlueScript plasmid. It should look something like this:
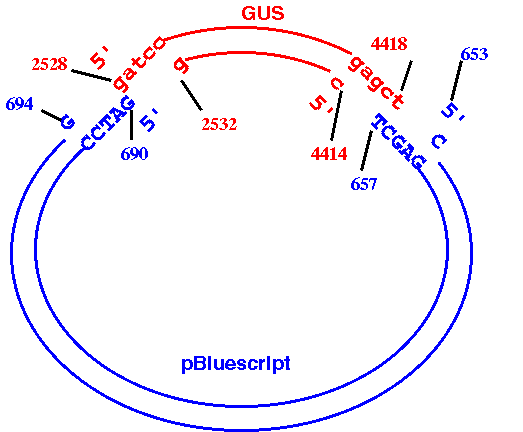
How to do it:
a. Read GenBank entries for pBI101 (PBI101TD.gen) and pBluescriptKSm13+ (X52331.gen) into bldna.
b. Use DNARNA --> BACHREST to find the locations of the BamHI and SacI sites in PBI101TD. (PBI101TD BACHREST output). According to BACHREST, the 5' ends of the BamHI and SacI sites are at 2528 and 4419, respectively. Therefore, the 3' end of the fragment we want is at 4418, not 4419.
(See 'II. What the output means' in the BACHREST documentation file rest.txt for details on the output.)
c. For PI101TD, use DNARNA--> NUMSEQ
START: 2528
FINISH: 4418
Save this output as pBSGUS.dna, and minimize the window to get it out of the way.
d. Use DNARNA --> BACHREST to find the locations of the BamHI and SacI sites in X52331. (X52331 BACHREST output). According to BACHREST, the 5' ends of the BamHI and SacI sites are at 690 and 658, respectively. Since the GUS fragment terminates at a SacI site, the SacI site from the plasmid must come next, with the BamHI site at the other end. We need to generate the opposite strand of the plasmid, going from the 5' end of SacI to the 3' end of BamHI.
e. For X52331, use DNARNA --> NUMSEQ
START: 653
FINISH: 694
WHICH: Opposite
Copy this output to the end of pBSGUS.dna and save the fille.
|
Have you adequately documented your work? One of the standards in scientific publications, and in all aspects of experimentation, is do document your work succinctly, but with enough detail so that any other worker skilled in the field would be able to reproduce what you did. Therefore, it's not sufficient just to say something like, Âthe GUS gene from pBI101 was cloned into Bluescript. Which Bluescript vector? Which restriction fragment, and which site or sites in the vector? See the sample file, pBSGUS.dna for an example that uses the GenBank accession numbers and feature syntax to precisely describe how the insert was cloned into the vector. |
f. Before going any further, verify that the construct has been built correctly. One way to do this is to use NUMSEQ to generate double-stranded printouts of both original sequences, and then mark the positions of the restriction sites on these printouts. Print out pBSGUS.dna and compare the sequence at the cloning junctions to the sequences in the originals. Make sure that complete BamHI and SacI sites appear at these junctions. There should be a BamHI site (G^GATCC) at 4809 and a SacI site (GAGCT^C) at 1892.
g. Convert pBSGUS into a Pearson/Fasta file.
In bldna, use File --> Import Free Format (FSAP), to read in 'pBSGUS.dna'. Note that free-format files do not contain sequence names, so the filename is used as the sequence name, in bioLegato. Since we don't want the .seq extension to be part of the name, get rid of '.dna' in Edit --> GetInfo. The name should now be 'pBSGUS'. Also, in the GetInfo menu for pBSGUS, change the topology to Circular.
There are two ways to save the file in FASTA format. One is to select the sequence and choose File --> SaveAs. You will need to set the file format to FASTA, and specifying a file name eg. 'pBSGUS.fsa'.
The other way to save the file is by choosing File --> Export Foreign Format. Set the output format to "Pearson/Fasta", and type 'pBSGUS.fsa' in the "Save As?" box.
h. Test your GenBank file by reading it into bldna, and running BACHREST. Because FASTA files do not have a way to specify sequence topology, you will have to explicitly tell bldna that the sequence is circular. After you have read in pBSGUS.fsa, select pBSGUS and choose Edit --> GetInfo. Change the topology to Circular. Now run DNARNA --> BACHREST. The BACHREST output should show that pBSGUS is circular, and the BamHI and SacI sites at 1 and 1892, respectively (pBSGUS.bachrest).