
The discovery of techniques which produce banding patterns on chromosomes is one of the most significant developments in cytogenetics. These techniques told us that chromosomes have an identity and structure that is consistent in all cells within a species, which supported the then-new chromosome theory of inheritance. Staining made it possible to construct the first chromosome maps, and to distinguish between chromosomes of many different species. However, fixed chromosomes are not equivalent to chromosomes in vivo. Fixation does not extract DNA from chromosomes but it does extract histones and nonhistone proteins to varying degrees. The fixative may not always have a quantitative, reproducible effect on these proteins. The net result is that when you look at fixed chromosomes, you are looking at chromosomes that are chemically and structurally different from chromosomes in living cells.
Some relevant terms to know when talking about chromosome banding:
Facultative heterochromatin often contains genes that are used
only at certain times and during certain cell activities, but
throughout the lifespan. Constitutive heterochromatin often
contains genes that are only used during early development and
remain tightly coiled and unused for the rest of that organism's
lifespan.
| Being able to match chromosomes by their band patterns is very useful, and assists with matching chromosomes under the microscope as well as classifying different areas of one chromosome. We produce these banding patterns (or G-banding) by using Giemsa stain. What G-banding attempts to do is to accentuate the differences in high-level chromatin structure along the length of the chromosome. In other words, some regions of the mitotic chromosome are very densely coiled (heterochromatin) and others are less densely coiled (euchromatin). G-banding experimental protocols vary depending on species. |
|
Most staining techniques have to be optimized for a particluar tissue in a particular species. Here is one example of a G-banding protocol for humans:
This procedure assumes that cells
have already been spread on slides and have undergone
pretreatment and fixation.
| When we see bands on fully-condensed mitotic
chromosomes, we know that they have coiled differently. Dark
bands are densely coiled, light bands are lightly coiled. If you look at the rightmost diagram, you can see that prometaphase bands combine as the chromosomes condense further. |
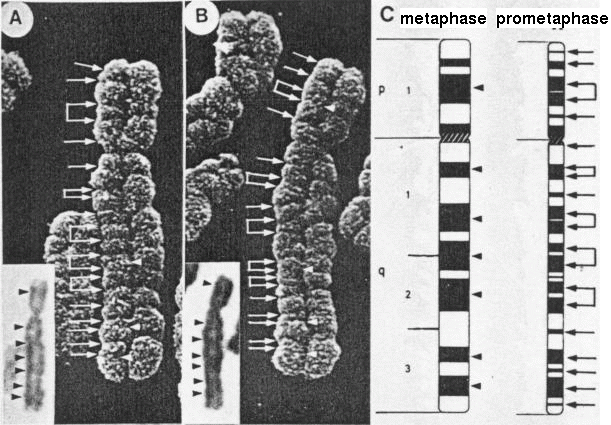 Figure 7. Composite image of a homologous pair
of slightly elongated number 5 chromosomes, with the
ISCN diagrams of metaphase (i) and prometaphase (ii)
chromosome band positions, and light micrographs of the
same chromosomes as insets. The major C bands in the LM
pictures are consistent with the metaphase band
positions in the diagram. Direct comparison of the
grooves seen in the SEM and the prometaphase diagram is
apparent. (Magnification x 10,000.) (Harrison, C.J.,
Jack, E.M. Allen, T.D., and Harris, R., J. Cell Sci.,
77, 143, 1985.) Figure 7. Composite image of a homologous pair
of slightly elongated number 5 chromosomes, with the
ISCN diagrams of metaphase (i) and prometaphase (ii)
chromosome band positions, and light micrographs of the
same chromosomes as insets. The major C bands in the LM
pictures are consistent with the metaphase band
positions in the diagram. Direct comparison of the
grooves seen in the SEM and the prometaphase diagram is
apparent. (Magnification x 10,000.) (Harrison, C.J.,
Jack, E.M. Allen, T.D., and Harris, R., J. Cell Sci.,
77, 143, 1985.)Accessed through: Creative Commons Attribution Non Commercial Share Alike License (the terms of which are set out at http://creativecommons.org/licenses/by-nc-sa/3.0/legalcode). |
Generally, features of chromosomes do not vary from tissue to tissue or change during the course of development. The organization reflected in banding is not random. In other words, we see consistent bands because the chromosome coils the same way every time.
| Figure 5. Low-power micrograph of a G-banded metaphase spread. The chromatid arms are segmented by circumferential grooves which run as a helix down each chromatid. (Magnification x 2800). | 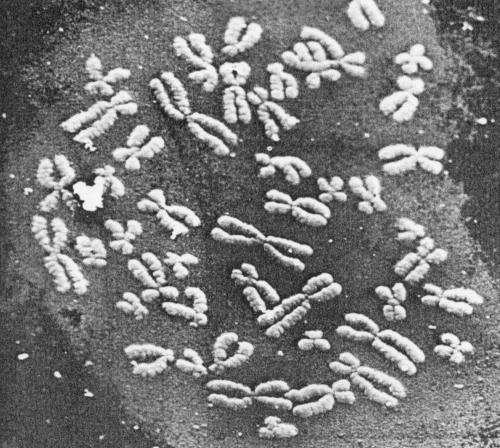 |
Figure 3. Part of a metaphase spread preparation prepared without banding techniques, illustrating small circumferential grooves on the surface of the chromatids. (Magnification x 9500.) |
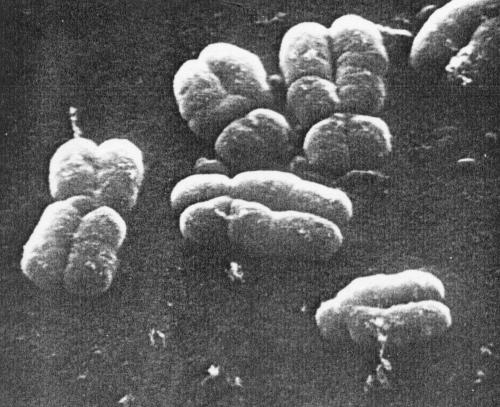 |
In specialized cells of some plant and animal species,
chromosomes undergo many cycles of DNA replication without
undergoing mitosis. The result is polytene chromosomes,
which may contain as many as a thousand parallel copies of each
chromosome, perfectly aligned along their entire length. Polytene
chromosomes can be viewed without
staining in preparations from cells in interphase.
The most common example is the polytene chromosomes in the
salivary glands of Drosophila. Since interphase
chromosomes are more extended than those in mitotic cells, loci
can be identified on polytene chromosomes at a much finer level of
resolution. Here is a drawing of one such polytene chromosome:
| Compare the size of polytene
chromosomes with normal mitotic chromosomes, shown
to scale in the inset! Drawing of polytene chromosomes in one Drosophila. Each chromosome is tightly paired with its homologue so that each pair appears as a single structure which is not the case in most interphase nuclei. The four chromosome pairs are linked together by regions near their centromeres that have aggregated to create a single large "chromocenter". The chromocenter has been split into two halves by the squashing procedure used. Note that the exact side-by-side alignment of many chromatin strands has greatly uncoiled each DNA molecule compared to its packing in a normal mitotic chromosome. This is an exceptional case of somatic pairing of homologous chromosomes. |
 Displayed by hypertext link to http://www.ncbi.nlm.nih.gov/books/NBK26847/bin/ch4f38.jpg |
As with Geimsa banding, chromosomes
must be fixed before denaturation. It is necessary to denature
both the DNA probe and the target chromosome to allow
complementary pairing to take place.
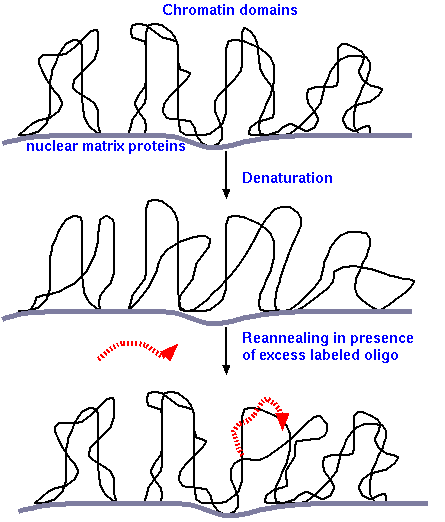
Fixative: The chromosomes are fixed in a 3:1 methanol:acetic acid solution. Slides are prepared by the squash method and the cover glass is removed to allow the cells to dry. The squash method stabilizes the chromosomes, permits good penetration of the reagents and brings most of the chromosome into the same focal plane, and also increases the speed at which the hybridization takes place, by decreasing volume.
Denaturation : incubate at 72° C in 50-70% formamide and 2X SSC (NaCl) (formamide denatures DNA helices). DNA remains covalently attached to nuclear matrix proteins only at Matrix Attachment Regions (MAR). Each loop of DNA between MAR sites is referred to as a chromatin domain. All other parts of the DNA helix are now single-stranded.
Annealing of probes and renaturation: The probe molecules are fragments of DNA, selected to target specific sequences on the chromosome. Probes can either be synthetic oligonucleotides (eg. 18 nt) or larger sequences of DNA. The target sequences can be as small as 1kbp or larger than 15 kbp.
Denatured probe is hybridized with fixed chromosomes under a coverslip overnight at 37° C in 50% formamide, 2X SSC and 10% detran sulfate. After incubation, unannealed probe is washed off . Because the probe is present in a large molar excess, compared to the chromosomal DNA, the probe will outcompete the single-stranded chromosomal DNA when base pairing with the strand complementary to the probe. Re-annealed duplexes are a hybrid of probe and native chromosomal DNA strands.
In summary, the chromosomes have been exposed to methanol, acetic acid, flattening, drying, rehydration, heat, formamide, and high salt concentrations during the FISH procedure. Some degree of structural changes will have been produced in the chromosomes. High resolution microscopic equipment is required for FISH as well as image processing software to enhance and analyse the image.
FISH is used to study genome organization, the distribution of chromatin in interphase nuclei, and chromosome abnormalities. Greater resolution is possible in interphase chromosomes due to the dispersed state of the chromatin, so that small structural changes can be detected. The compaction of metaphase chromosomes sets the limit of detection at sequences separated by more than 1 Mbp. The limit in interphase chromosomes is a separation of 100 kbp (Figure 8, Trask et al. 1993).
| When the probe is specific for a single gene, only that gene is expected to light up in FISH. The image shows hybridization of a probe for the neu proto-oncogene in human lymphocytes. The neu locus resides on chromosome 17. Since the cells contain diploid nuclei, two hybridizing spots are seen in each nucleus. The sensitivity of FISH is put into perspecive by considering the fact that each chromosome is a single DNA molecule. That is, each spot results from a few probe molecules hybridizing to different parts of a single DNA molecule. Looked at another way, we can recall that Avogadro's number (the number of molecules in a mole) is 6.02 x 1023 molecules/mole, we are detecting 1.66 x 10-24 moles of chromosomal DNA! |
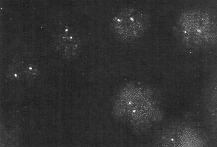 |
| It is possible to isolate DNA specifically
from individual chromosomes. When total chromosomal DNA is
used as a probe, it will base pair with all sequences on the
chromosome from which it was derived, making the entire
chromosome 'light up'. In other words, each sequence in the
probe eventually encounters its complementary sequence on
the chromosome and base-pairs with it. M-FISH
or multicoloured FISH is particularly useful for identifying
individual chromosomes in a karyotype, as well as any
genetic abnormalities. With M-FISH, each chromosome is
painted a different colour. |
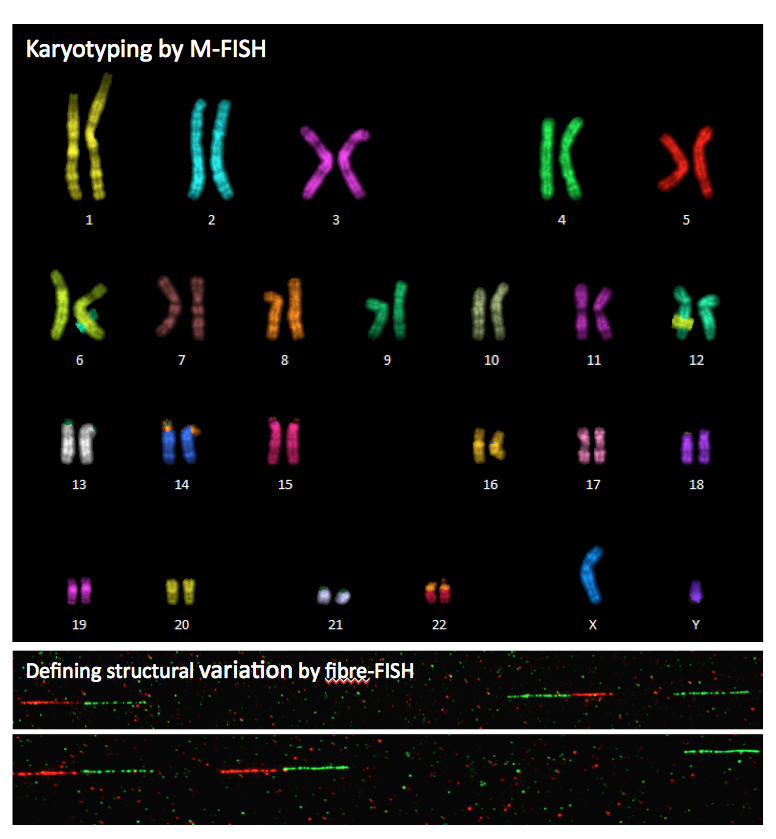 From Sanger
Institute, Molecular Cytogenetics From Sanger
Institute, Molecular Cytogenetics |
| M-FISH has also revealed that chromosomes tend to occupy certain parts of the nucleus referred to as 'territories'. The existence of chromosome territories reveals a fundamental level of organization in the interphase nucleus. It implies that as chromosomes decondense, rather than randomly unspooling their chromatin strands, a more orderly unpacking must occur, probably concurrent with the re-establishment of the nuclear matrix. |