When compatible ends are in close proximity, DNA ligase can create phosphodiester bonds between the 5' and 3' ends of two fragments, re-creating the restriction site.
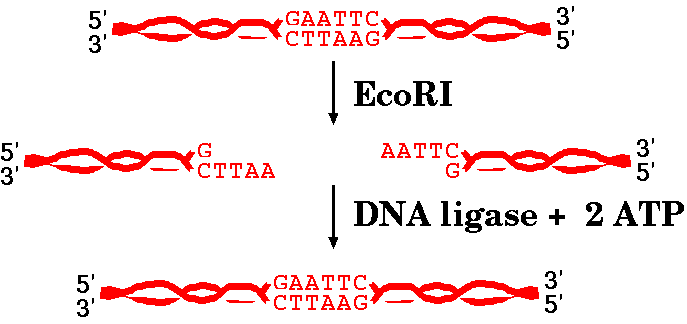
DNA ligases are enzymes that are capable of joining different pieces of DNA together. You may remember that DNA ligases play a role in DNA replication, by joining the Okazaki fragments of lagging strand synthesis together.
| We discussed in the last reading that EcoRI
produces "sticky ends", or ends that have an overhang and
can spontaneously base pair again. When compatible ends are in close proximity, DNA ligase can create phosphodiester bonds between the 5' and 3' ends of two fragments, re-creating the restriction site. |
|
There are also restriction endonucleases that produce "blunt ends", like SmaI, which has the recognition sequence CCCGGG.
5' CCCGGG 3'
3' GGGCCC 5'
digest with SmaI
5' CCC 3' 5' GGG 3'
3' GGG 5' 3' CCC 5'
Not all restriction endonucleases create fragments with complementary ends, and sometime different fragments can create complementary ends for each other. The takeaway from all this is that sometimes ends can spontaneously base-pair again and sometimes not. However, base pairing is not enough for the DNA strands to be whole again. DNA ligase has to seal the sugar-phosphate backbone together.
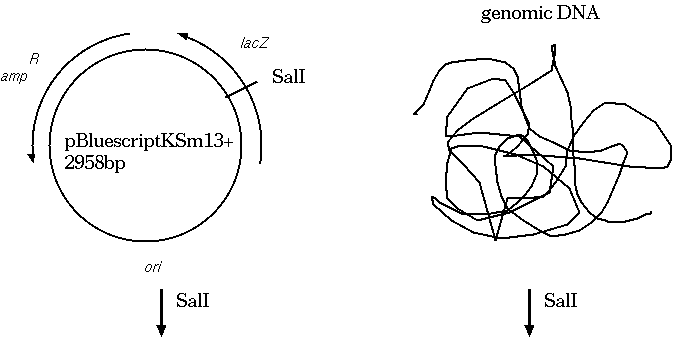
Restriction endonucleases and DNA ligases give us the necessary
means to clone genetic sequences from almost any
organism into a plasmid vector. Here's how it works: We digest
both the target DNA and the plasmid with the same
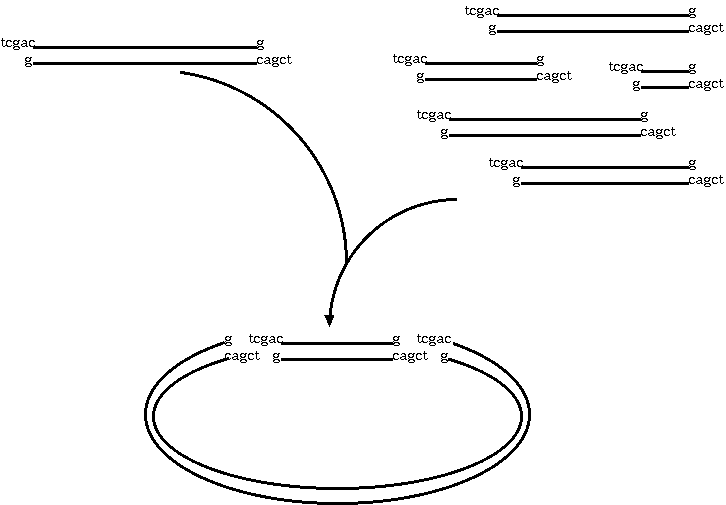
restriction endonuclease. Let's say we used SalI. We're going to
end up with overhang, "sticky" ends, which will automatically
re-anneal. At many of the sites, the plasmid DNA will re-anneal
with itself, but some will re-anneal with the other genetic
material. What we end up with is plasmids that contain a portion
of our target DNA. Usually, the plasmids are manipulated in other
ways so that we can tell which plasmids have the target DNA.
A typical cloning experiment follows this outline:
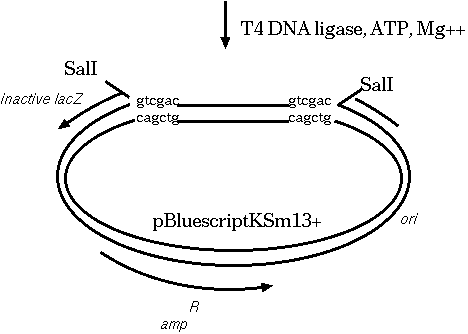
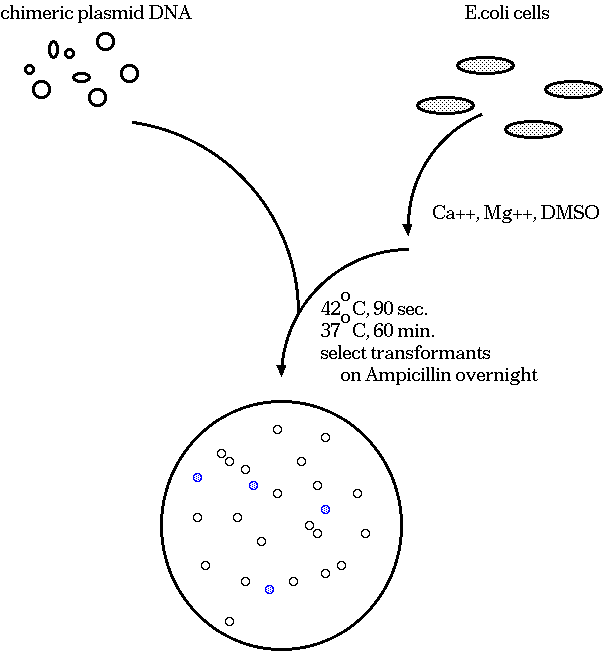
Clones that survive must have the plasmid, because they are
ampicillin resistant. White colonies have lost β-glactosidase
activity, indicating that a fragment of target DNA has inserted
within the lacZ coding sequence. Therefore, we pick white
colonies, each of which is a presumptive clone containing an
insert.
| A synthetic DNA sequence has been inserted downstream from the translation start site for the lacZ gene. Translation begins at position 816 with the ATG codon. The multiple cloning site, going from KpnI to SacI, is designed to have many convenient restriction sites, making it possible to clone a restriction fragment generated by almost any restriction enzyme somewhere in the MCS. The inserted fragment has a length divisible by 3, and sequences have been chosen so that there are no stop codons within the MCS. Although the lacZ protein will contain a few extra amino acids in its N-terminal region, these extra amino acids do not interfere with its enzymatic activity. | 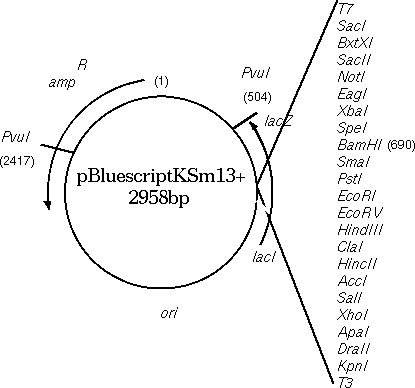 |
The sequence of the multiple cloning region shows that the lacZ
gene in the Bluescript vector has been engineered to contain
artificially-introduced restriction sites, to give us a choice of
sites for insertion of fragments.
m13 reverse primer: aacagct SalI ClaI HindIII EcoRV EcoRI PstI SmaI BamHI SpeI XbaI NotI |
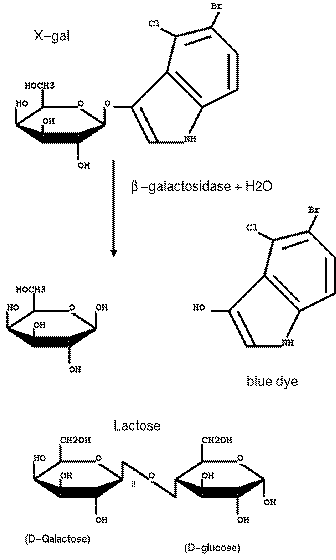
| The multiple cloning
site was carefully crafted so that no stop codons were
inserted, and that the reading frame of the lacZ gene
was conserved. The result is that the N-terminus of the
β-glactosidase protein has some additional amino acids
at its N-terminus. However, the enzyme is still
functional even with these additional amino acids. X-GAL CLEAVAGE BY beta-GALACTOSIDASE X-gal : 5-bromo-4-chloro-3indolyl-beta-D-galactoside. |
 |
|
We've already seen with FISH that you can detect a DNA
sequence on a chromosome by hybridizing a labeled DNA
probe to chromsomes on a microscope slide. For identifying DNA from a gel, the DNA is first blotted
onto a nylon membrane, and incubated with a labeled DNA
probe. (We'll discuss gel blotting below).
|
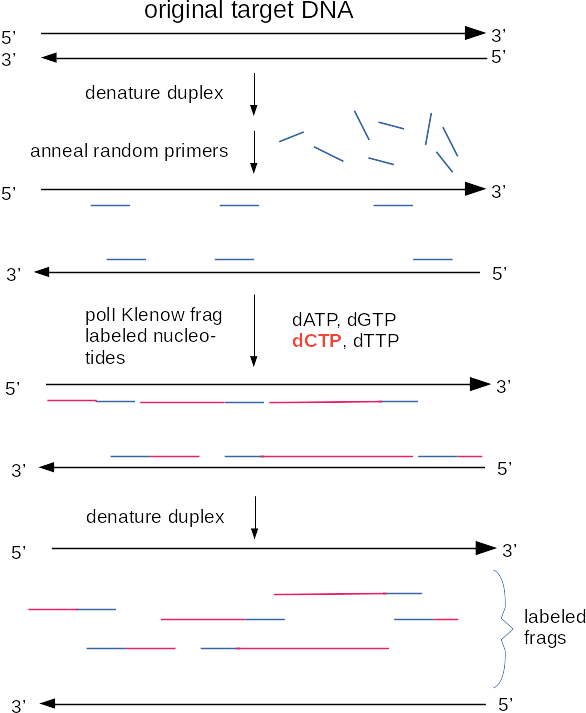 |
Another method for making labelled probes uses PCR. PCR, or the polymerase chain reaction, is a method for amplifying segments of DNA [PCR Review]. The DNA labelling reaction using PCR is similar to the regular PCR reaction, except labelled nucleotides are incorporated with the unlabelled nucleotides. Some advantages of choosing to label DNA this way are:
Although radioisotopes were used for probe synthesis at one time, radioisotopes have been replaced by two general approaches to non-radioactive labeling. Both of these methods incorporate various nucleotide analogs into DNA in a labeling reaction. In chemiluminescence, an enzyme is conjugated to a molecule such as an antibody that is specific for the nucleotide analog. When the appropriate substrate is added, the enzyme breaks it down, resulting in the emission of a photon of light. In fluorescent detection, nucleotide analogs themselves contain a fluorescent tag, which emits light when illuminated with the appropriate excitation wavelength. Commonly used nucleotide analogs include: Dioxygenin-labelled nucleotides (DIG-dNTPs), biotin-labelled nucleotides, and fluorescent nucleotides. Each of these types of nucleotide analogs is detected differently.
| Name | Detection Method | Example |
|---|---|---|
| DIG-dNTPs | Detection of DIG-labeled nucleotides is done using anti-DIG antibodies conjugated to alkaline phosphatase. When a substrate such as CDP* is added, alkaline phosphatase breaks down the substrate, which emits a photon of light. | 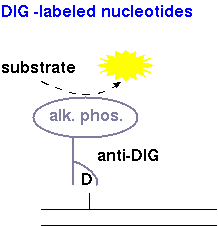 |
| Biotinylated DNA | Detection of biotinylated DNA is typically done using streptavidin, a bacterial toxin that has an affinity for biotin, conjugated to horseradish peroxidase. Breakdown of substrate (eg. luminol peroxide from Clontech Inc.) results in release of a photon of light. | 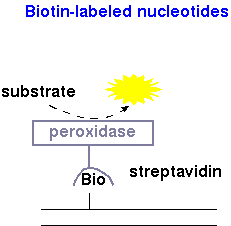 |
| Fluorescent DNA | Fluorescently-tagged DNA probes have the advantage that no enzymatic reaction is necessary for detection. Dye-conjugated nucleotides are incorporated into DNA during labeling eg. PCR labeling. | 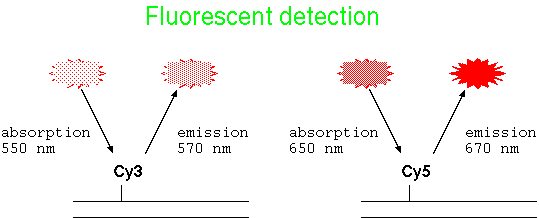 |
The purpose of hybridization is to use a labelled sequence of DNA (called a probe) to detect a specific, complementary sequence in a genome. Hybridization has many uses, including:
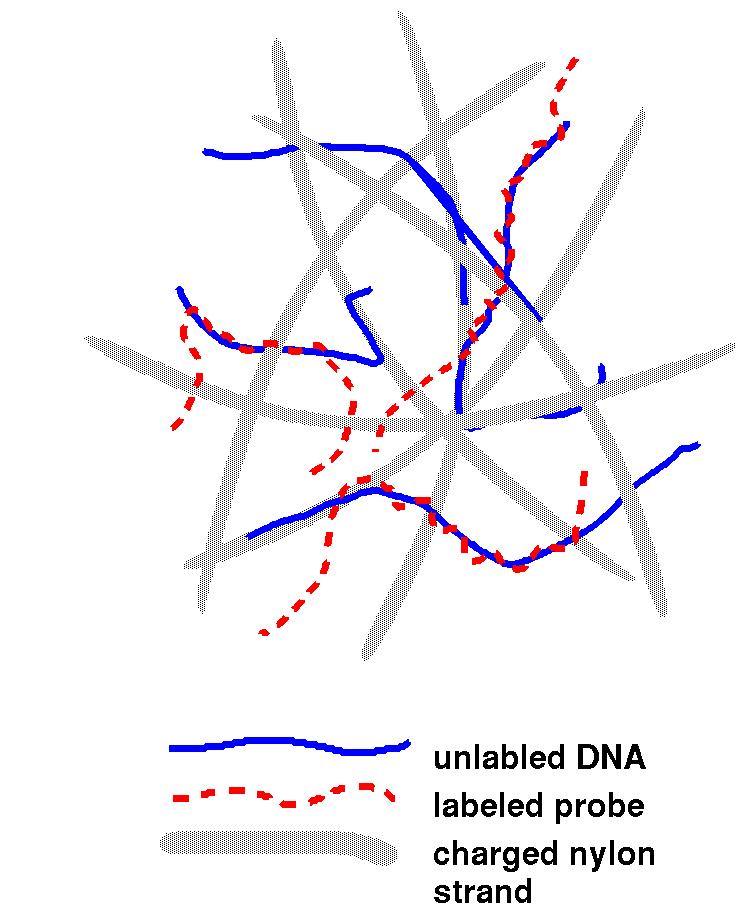
| The image at right is a representation of the
filter (grey) used in hybridization experiments. Once the
unlabelled DNA is denatured (with an alkaline solution like
NaOH), the transfer is made at neutral pH to keep the DNA
mostly in single-stranded form. The DNA then adheres to the
filter strands. The probe is denatured as well, and combined
with the filter to incubate. Then, the probe is free to base
pair with complementary regions. After sufficient time has
passed, washing is used to clean excess unhybridized probe
from the filter. On the filter, there is little opportunity
for complementary strands to find each other, so most DNA
remains single-stranded. |
 |
By blotting the DNA from the gel to a nylon filter, the DNA bands
are transferred in the same pattern in which they occur on the
gel. Now, if we incubate the filter with the labeled DNA
probe, the probe will bind to only those bands that have a
sequence complementary to the probe. Even imperfect matches
between the probe and the target DNA will result in a stable
duplex, which we detect as signal on the filter.
Detection is done either using fluorescence or chemiluminescence,
as described previously.
See: SCHEMATIC OF HYBRIDIZATION EXPERIMENT
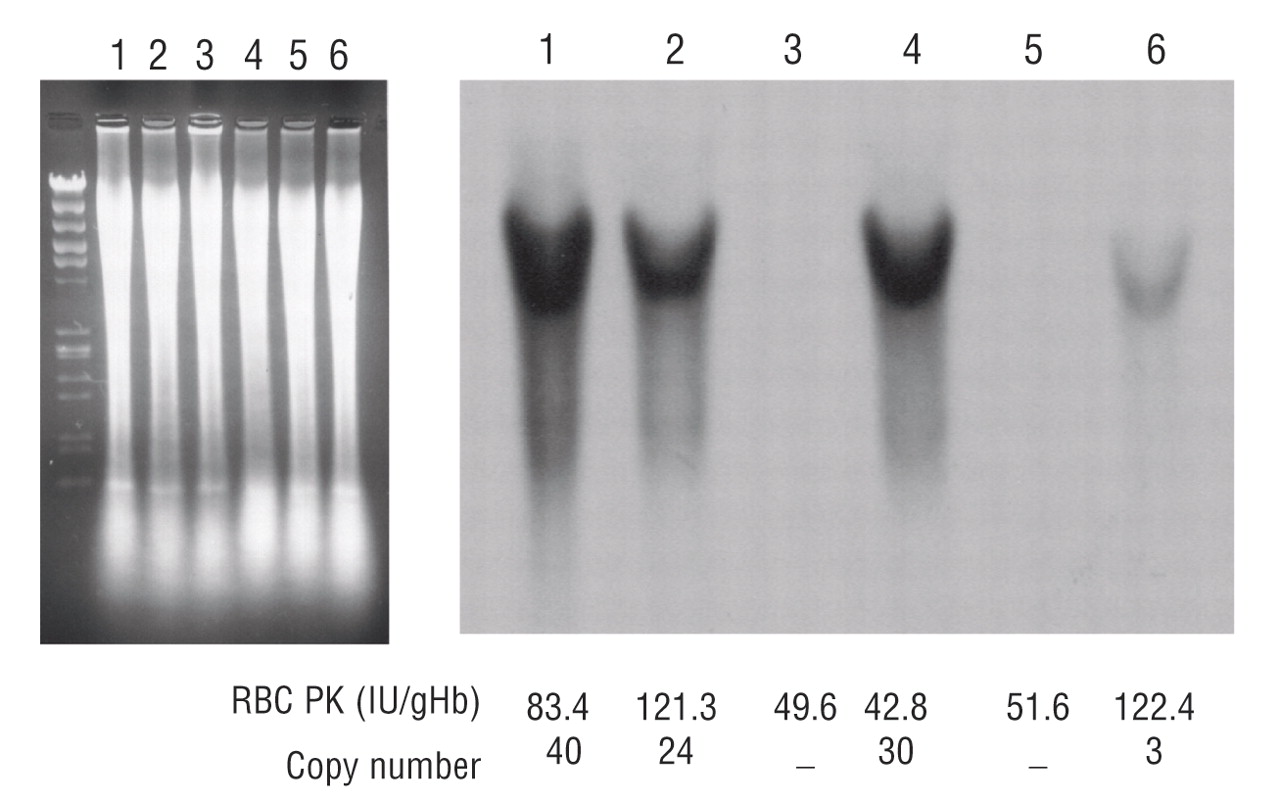
Transgenic rescue of hemolytic anemia due to red blood cell pyruvate kinase deficiency
Hitoshi Kanno, Taiju Utsugisawa, Shin Aizawa, Tsutomu Koizumi, Ken-ichi Aisaki, Takako Hamada, Hiromi Ogura, Hisaichi Fujii
Haematologica June 2007 92: 731-737; doi:10.3324/haematol.10945
DNA sequencing methods have come a long way since the early
1990s. The methods below are all "next generation sequencing" or
NGS methods. The key feature of NGS methods is that they are able
to sequence thousands or millions of short DNA fragments at a
time, drastically reducing the per bp cost of sequencing. The
types of sequencing differ depending on whether you need to
sequence an unknown genome (de novo), or resequence an
already-known genome.
All of these methods share a common strategy:
1. Cut genomic DNA into millions of short fragments
2. Sequence millions of fragments
3. Use genome assembly software to put together overlapping
sequencing reads into contigs. Contigs are large fragments of
chromosomes
Pyrosequencing uses pyrophosphate as a indicator. When a dNTP is added, pyrophosphate is released and through a cascade, cleaves a substrate called luciferin, which then emits a photon. Only 1 of the 4 dNTPs is only added in each cycle, so if a light signal is detected you know which nucleotide was added. This process produces reads of around 500 bp.
This video explains more of the process than the one originally included below. New:Original:
Illumina sequencing uses dNTPs that all fluoresce at 4 different
wavelengths. Fragments of DNA are tethered to a flowcell, and
amplified so that many copies of each fragment can be used. Then,
when each nucleotide is added, that particular area on the
flowcell flashes a distinctive colour for each different
nucleotide. Current Illumina platforms can read up to 250 bases
per read.
Single molecule real-time sequencing, or SMRT, is a technology developed by the company Pacific Biotechnology. This technology also uses differently-labelled nucleotides, but they are attached to the terminal phosphate instead of to the sugar. SMRT uses an extremely small detection chamber to catch the flashes of different coloured light to read the sequence of the DNA. The advantage of this method is that it produces much longer read lengths, to about 20,000 bp. However, it has a high error rate.
These technologies all produce sequences, or "reads", only of a certain length. In order to assemble these reads into a continuous genome, we need different tools.