TUTORIAL: Genome
Assembly
|
December 1, 2023 |
TUTORIAL: Genome
Assembly
|
December 1, 2023 |
| We want to assemble a genome
using the three pairs of corrected read files. |
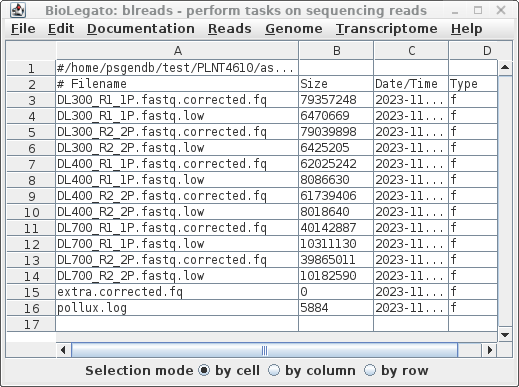 |
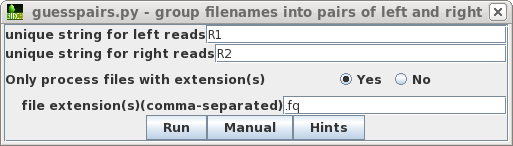
| Choose File -->
guesspairs. As before, we use R1 and R2 to identify paired-end read files, and ask blreads to only select files with the .fq file extension. |
 |
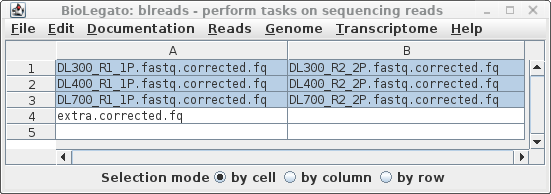
| A new blreads window appears
with the paired-end files in two columns. Since
extra.corrected.fq is empty, we won't bother including
that file. Select all pairs of reads, and choose Genome
--> ABySS. |
 |
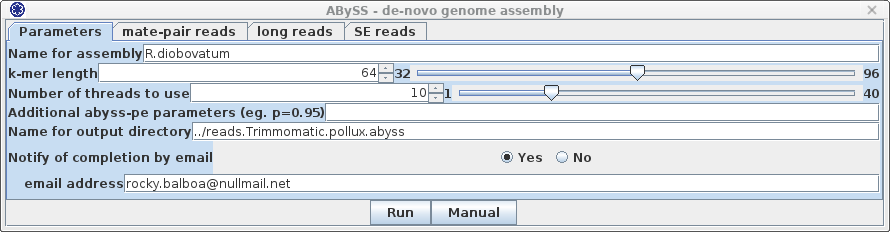
| It is a good idea to specify
a descriptive name for this assembly, in this case R.diobovatum.
By default, output will be written to a directory called
../reads.trimmed.corrected.abyss. Again, we want to be
more precise, so let's call it ../reads.Trimmomatic.pollux.abyss.
Since it takes a long time to do an assembly, it is best
to set Notify of completion by email to Yes, and type in
your email address. ABySS has a large number of parameters, not all of which are implemented in blreads. You can set additional abyss parameters, as described in the abyss manual page. If you had additional files with mate-pair reads to be used for scaffolding, those can be added in the mate-pair tab, shown at right. |
  |
| As described in the ABySS
documentation, the final genome assembly is found in
R.diobovatum-contigs.fa and R.diobovatum-scaffolds.fa.
(These are actually symbolic links to the final contigs in
step 6, and the scaffolds in step 8). To see the summary of results, select abyss.log and choose File --> View file. |
 |
| The statistics of the final
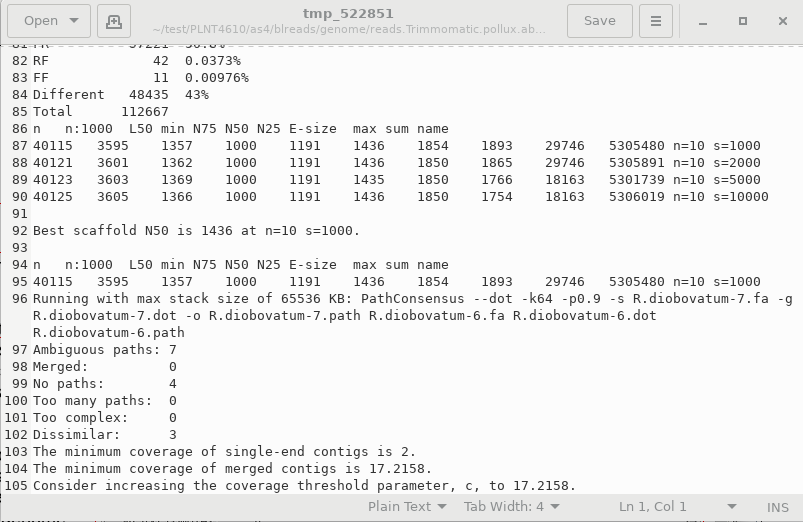
assembly are shown at the bottom of this file. We see that the number of contigs is 40115, and the median size of scaffold, N50 is 1357 bp. The longest scaffold was 29746 bp. The total size of was about 5.3 million bases. These results would not be good if we were working with a complete set of reads. This test dataset does not have the coverage to give a high quality genome. Since the R. diobovatum genome is over 21 million bases long, less than 1/4 th of the genome is represented in the final scaffolds. |
 |
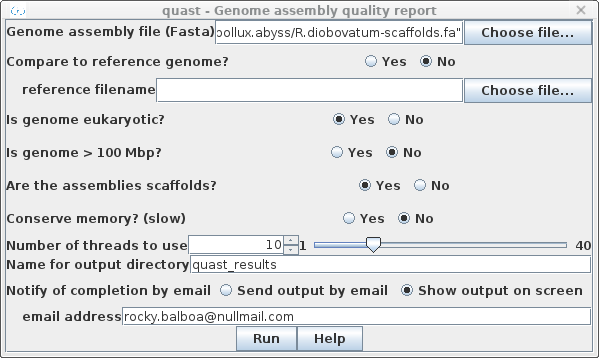
| Choose Genome -->
Quast. The genome assembly file could either be the contig file, R.diobovatum-contigs.fa, or the scaffolds file, R.diobovatum-scaffolds.fa. The latter us normally preferable. Since this genome is from a fungus, set Is genome eukaryotic to Yes. As well, if the input is scaffolds, set Are the assemblies scaffolds? to yes. Unless your system has very limited memory, it is best to leave Conserve memory to the default setting of "No". By default, output is written to a directory called quast_results. |
 |
1) quast.log - the details of the quast run
2) report.html - a detailed report of quast statistics, including graphs
3) blreads - showing all files in the quast_results directory. Files can be selected and opened using File --> View file.
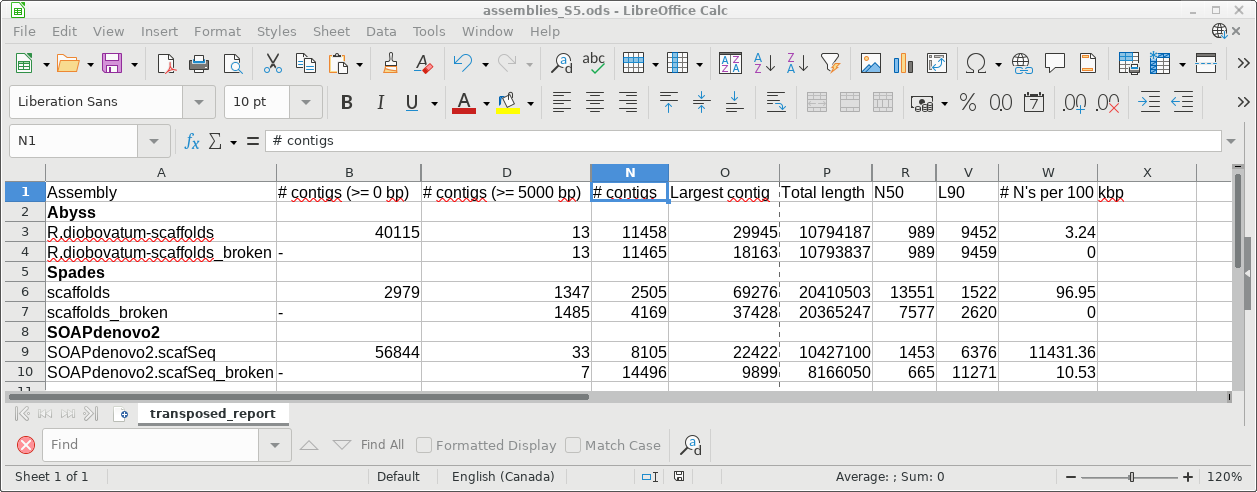
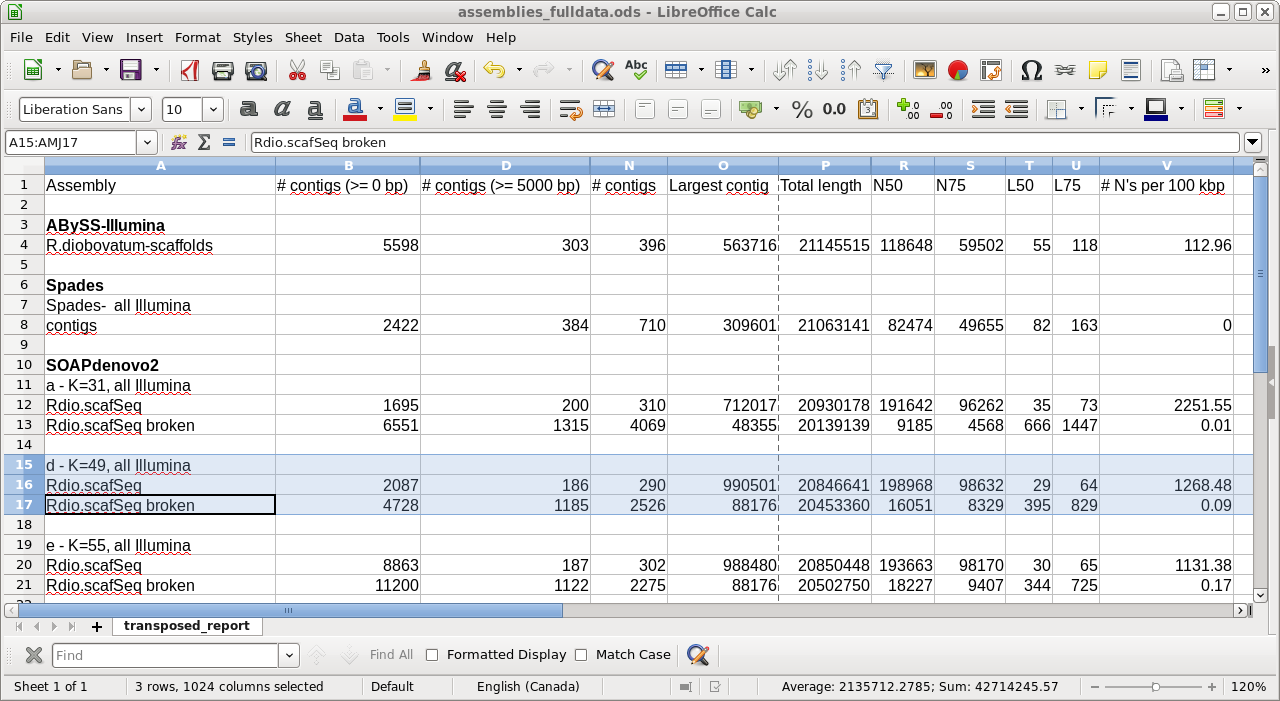
4) transposed_report.tsv - This file contains the same statistics as in report.html, but in spreadsheet form. This is actually a very useful file, because you can copy the lines from this spreadsheet into a master spreadsheet for different genome assemblies, making it easy to compare assemblies for each criterion in the spreadsheet. For example, in the next section, we will assemble the same genome using Spades, and then compare the results from both assemblies in a single spreadsheet.
| Spades has its own read corrector, and spades seem to construct better assemblies when read correction is done by spades, rather than pollux. Start blreads in the reads.Trimmomatic directory, and run guesspairs to select P.fastq files. |  |
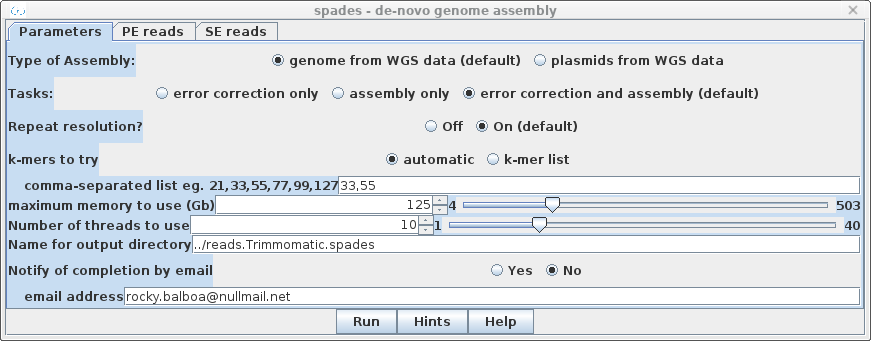
| By default, spades will do
error correction as the first step. Unless a k-mer list is
supplied under k-mers to try, spades will automatically
repeat the assembly with k-mers of sizes chosen by spades.
To document the steps done, set the name of the output directory to ../reads.Trimmomatic.spades. |
 |
| As described in the Spades
documentation, spades will try constructing the contigs
using several sizes of k-mers (21, 33, 55 and 77). The
final contigs will be found in contigs.fasta, and
scaffolds in scaffolds.fasta. Where mate pair sequences
are not available, there will not be a great deal of
difference between these two files. To see a summary of the results, select spades.log and choose File --> View file. |
 |
| The reads for the first
library, as well as global parameters, are set in the
Parameters tab. (For longer reads (eg. >250), max. k-mer size would be set to 127.) With this example, the best results were obtained setting k-mer size to 31, which is about 1/5 the size of the sequencing reads. The expected genome size is 21 Mb, so set Genome size to 24 Mb to make sure enough RAM is used. For eukaryotic genomes, it is always best to set "Use reads to resolve repeats" to Yes. In this example, the first library, LIB1, has left and right members chosen for Read Pair 1, with Avg. insert size set to 300 bp inserts. The Max. read length for these reads has been set to 150 nt. |
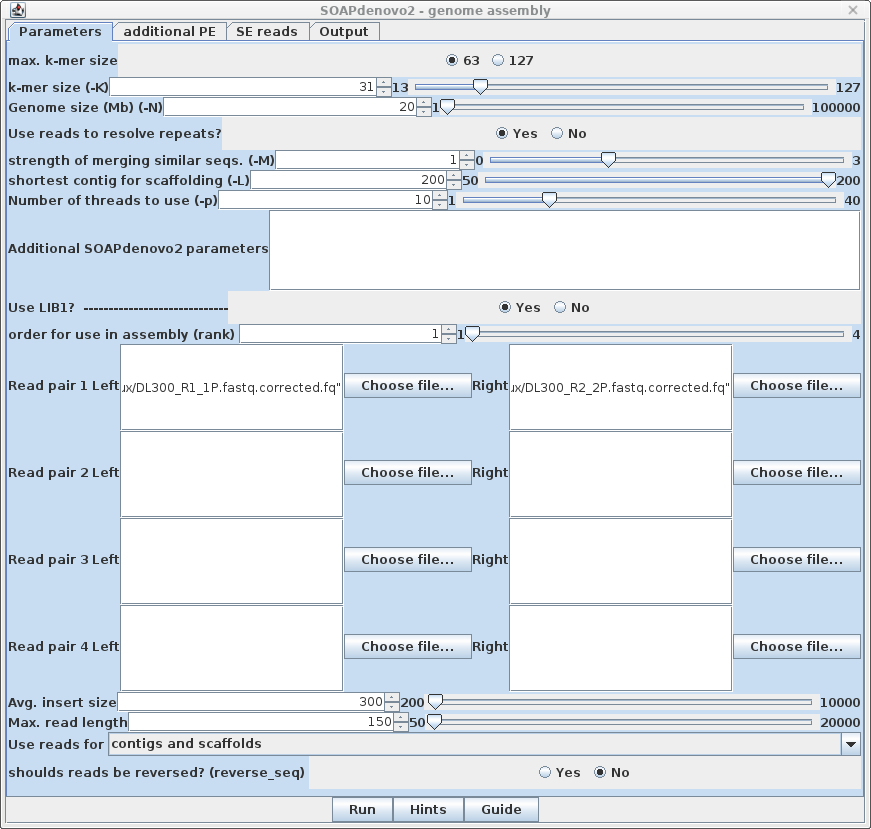 |
| This assembly also has files
with 400 and 700 bp inserts, so we specify these files and
their parameters in the AdditionalLIBS tab. For the 400 bp inserts, click Yes for Use LIB2 and set rank to 2. Set Avg. insert size to 400, and Max. read length to 150. Similarly, for the 700 bp inserts, click Yes for Use LIB3 and set rank to 3. Set Avg. insert size to 700 and Max. read length to 150. Note on mate pair reads: If you are using mate pair reads for scaffolding, put these in a separate library and set Use reads for scaffolds only. |
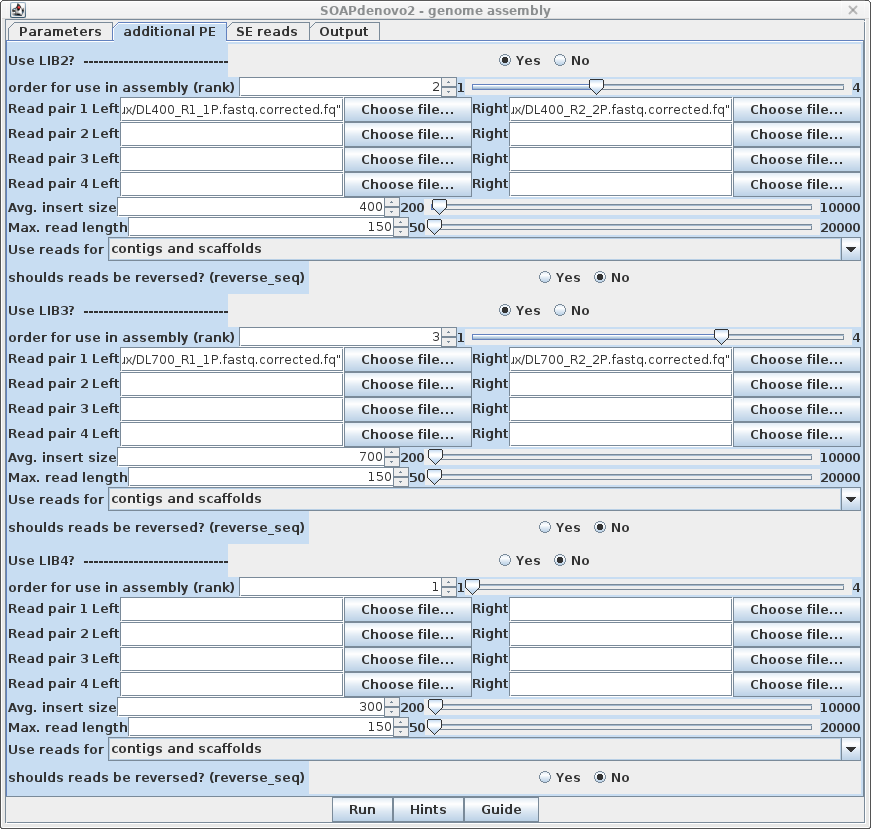 |
| An example of output as shown
in soapdenovo2.log, is shown at right. |
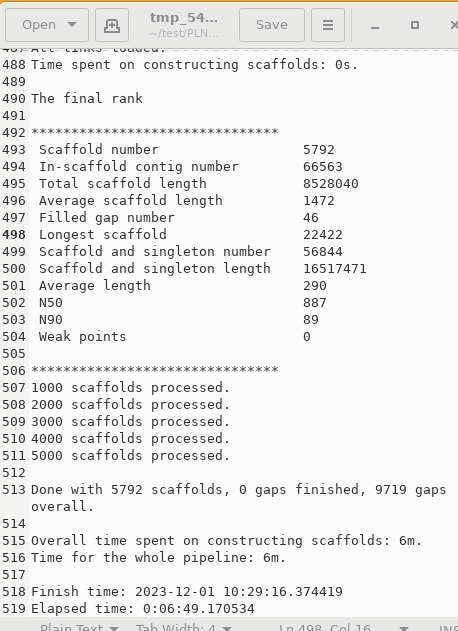 |
| Take home lesson: There is no substitute
for experimenting with different programs and a range of
parameters to obtain the optimal genome assembly. |