TUTORIAL:
Genome
Assembly
|
November 30, 2023 |
TUTORIAL:
Genome
Assembly
|
November 30, 2023 |
| Running pollux Your blreads window should now appear as shown at right. The read files will have the .fastq file extension. |
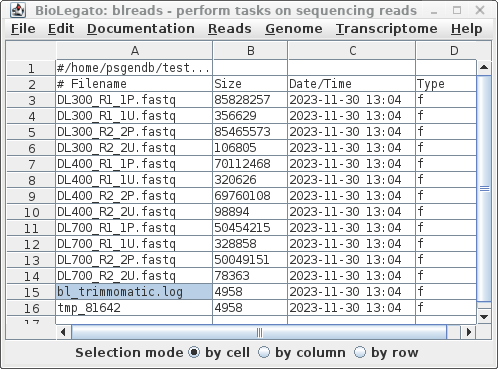 |
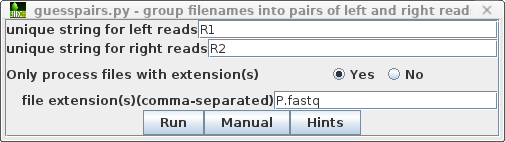
| We only want to process the paired read files
from Trimmomatic. Choose File --> Select All, and
then File --> guesspairs.py. Set the target file
extension to P.fastq. (In some cases, you may
need to specify more than one set of file extensions as a
comma-separated list eg. .fq,.fastq Clicking on the Hints
button will give a more detailed explanation of these
parameters.) |
 |
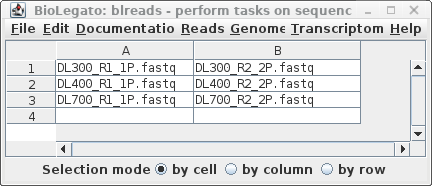
| Clicking on Run will bring up a new
blreads window with the best quess of file pairing, in two
columns. To run Pollux with these read pairs, choose Edit --> SelectAll, and then Reads --> Pollux. |
 |
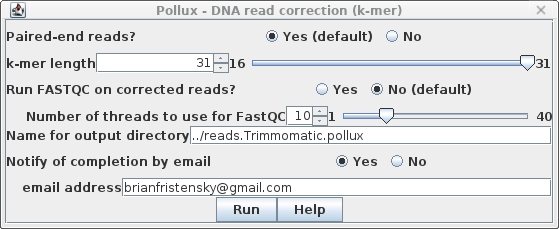
| Set the Name for output directory to
../reads.Trimmomatic.pollux. Remember that "../" will tell
BioLegato to write the output directory to the parent of the
current working directory. Pollux may take awhile to run, so you may wish to set "Notify of completion by email" to Yes and type in an email address. |
 |
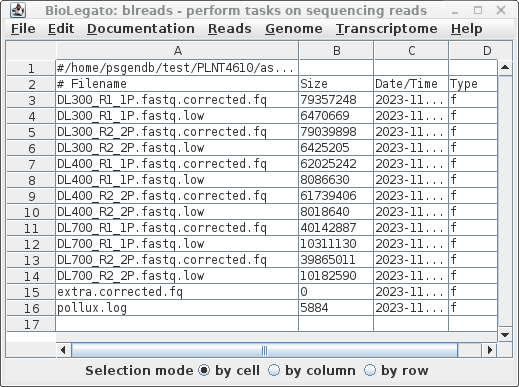
| The output is written to the
../reads.Trimmomatic.pollux directory, whose contents is
shown at right. The report in pollux.log summarizes the numbers of errors corrected of different types eg. insertions, deletions, corrections, homopolymer corrections. The high quality corrected reads are found in files with the .corrected.fq extension. Corrections based on low k-mer counts have the .low extension, and are generally not used. |
 |