TUTORIAL: DATA
PIPELINING WITH BioLegato
|
Nov. 8, 2021 |
TUTORIAL: DATA
PIPELINING WITH BioLegato
|
Nov. 8, 2021 |
As an example we’ll start with the amino acid sequence for the
SARS-Cov-2 Spike (S) protein, which is the best potential vaccine
target. Yuan and colleagues have used X-ray crystallography to determine
the 3D structure of the spike protein when complexed with a
neutralizing antibody from a patient infected with a SARS virus from
2007.
The authors demonstrated that decreased binding of the 2007 antibody to
the 2020 Spike protein could be explained by amino acid substitutions at
stearicly critical positions in the antibody binding site, compared to
the earlier spike protein from 2007.
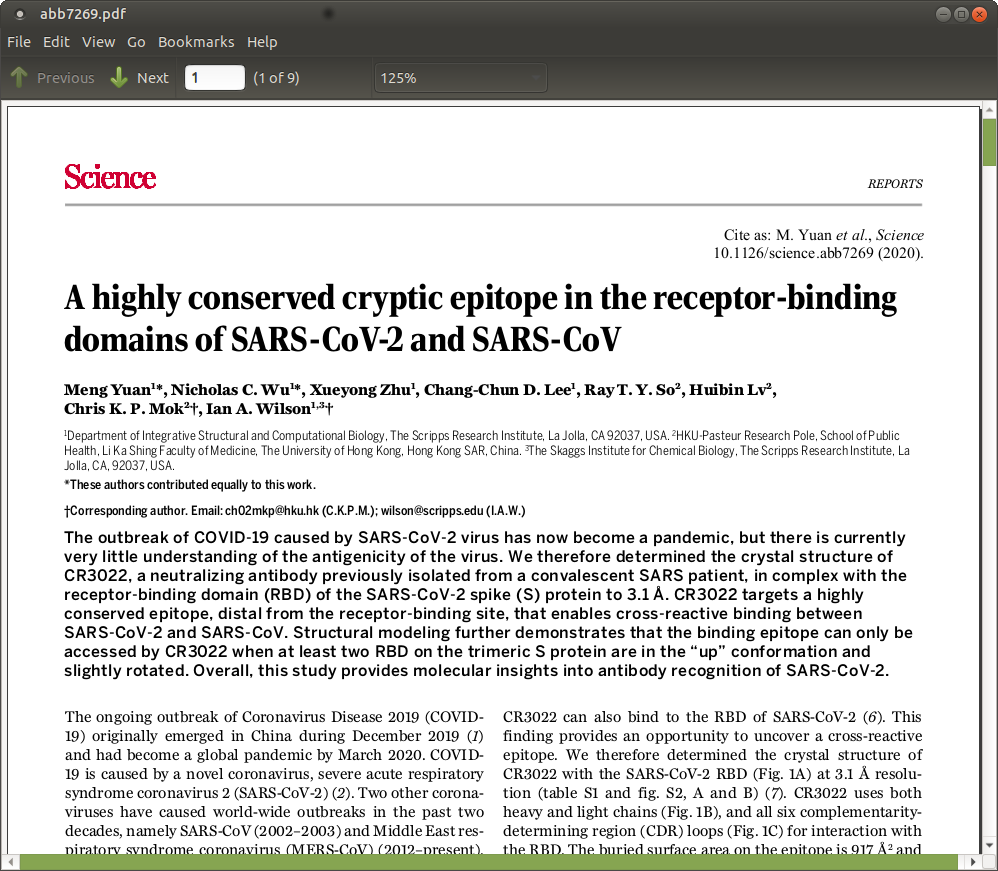
| In panel B we see the antibody heavy chain in
gold. The 2007 protein has an Alanine at position 372 creates an
N-glycosylation site at posn. 370. Mutation to a Threonine at 370
eliminates that N-glycosylation site (Fig. 2C) In this light, it would be instructive to compare Spike proteins from a number of SARS strains for a more thorough understanding of amino acid conservation at these positions. 2D - Antibody binding may also be affected by substitution of the Alanine at position 384 in the 2007 protein, with a Proline, which is likely to result in conformational changes. |
 |
| From an epidemiological perspective, it would be
useful to know whether these mutations are very recent, or whether they
have been present in distinct strains of the SARS virus for a long time. For that we need to compare our two spike proteins with spike proteins from other SARS strains in a multiple sequence alignment. The best way to find those sequences is to use the these spike proteins as queries in BLAST searches. |
 |
That means that first we have to retrieve the two proteins described
in this paper, using the accession numbers from the manuscript.
In your tutorials directory, create a
directory called 'pipeline' and go to the pipeline directory
cd tutorials
mkdir pipeline
cd pipeline
|
Launch blncbi in the background. blncbi & |
 |
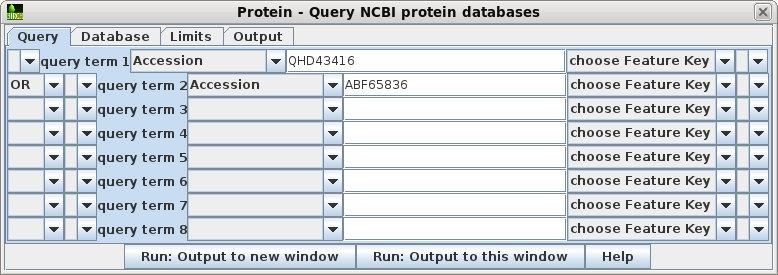
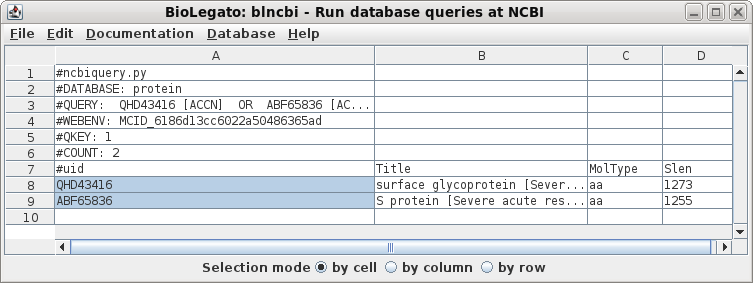
| Choose Database --> Protein. For the first query term, set the field to Accession, and the value to QHD43416 For query term 2, set OR, so that sequences matching either of the queries will be found. Set the field to Accession and the value to ABF65836. These settings will send the following Entrez search expression to NCBI: QHD43416 [Accession] OR ABF65836 [Accession] |
 |
| The results pop up in a new blncbi window. |
 |
| Choose Database --> PROTFETCH. Click on Run. The GenBank protein entries will be retrieved to a bldna object. |
 |
| The GenBank protein entries will be retrieved
to a blprotein object. |
 |
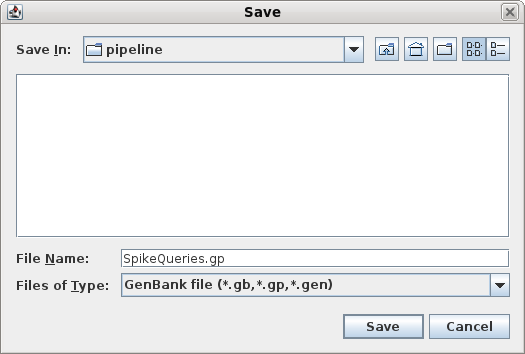
| To save these entries, choose File -->
Save ALL As Set the file name to SpikeQueries.pg and set the file type to GenBank. At any later time, you could open this file in blprotein. For now, it's worth having a look at the sequences. In the file manager, double click on the file to open it in a text editor. |
 |
| Choose Edit --> Select All and
then Protein --> Protein statistics. Set Output
to Table, and run the program. |
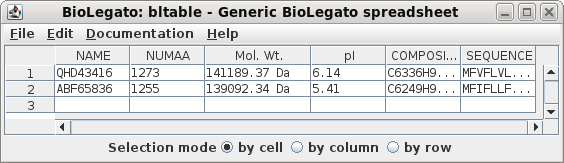
 |
| We can see that these are moderately sized
proteins, over 1200 amino acids each, or over 13 kDa in
molecular weight. Both are slightly acidic, with the 2020
protein being somewhat less acidic than that from 2007. |
 |
| In blprotein, choose Similarity -->
SSEARCH. SSEARCH performs a rigorus
Smith-Waterman alignment between the two proteins, resulting
in an optimal alignment. To evaluate the statistical
significance of the alignmet, SSEARCH will repeat the
alignment 500 times, randomizing the sequences in each
case. If the score of the optimal alignment is better than
alignments of randomized sequences, the optimal alignment is
considered better than an alignment of proteins of similar
length and amino acid composition, by random chance. |
 |
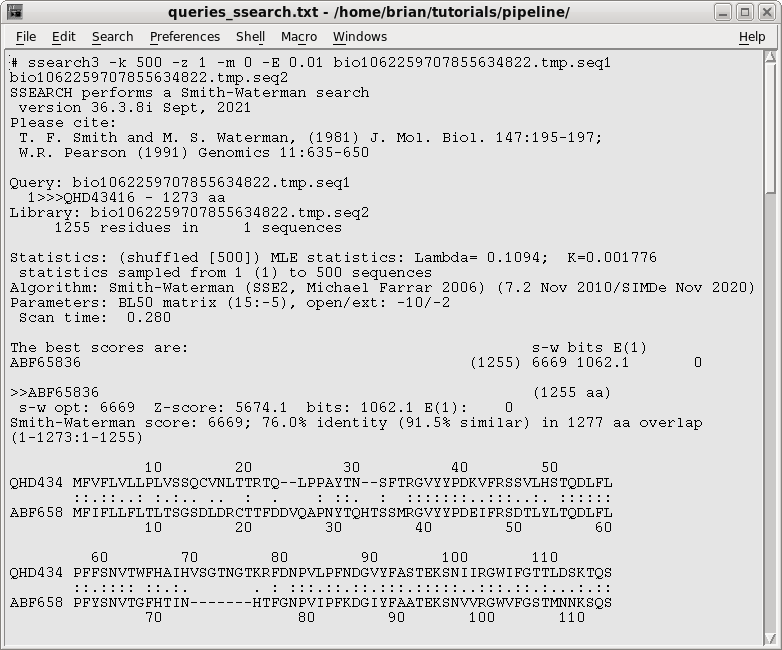
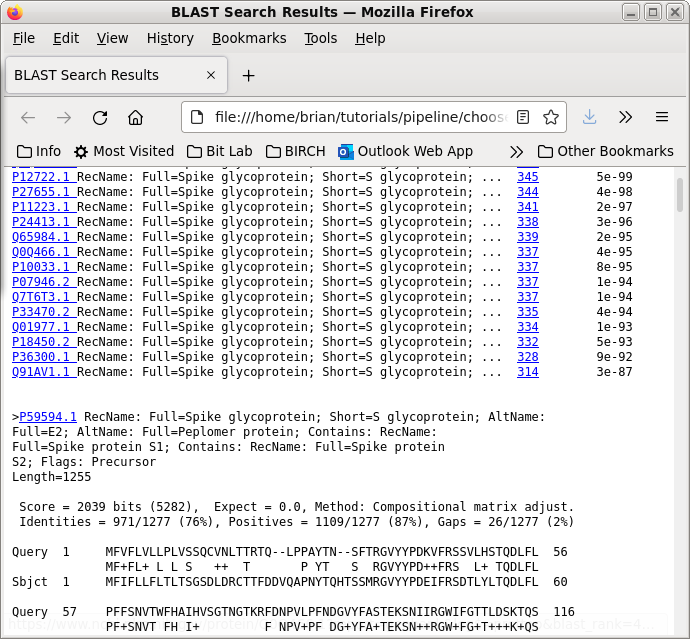
| A partial output is shown at right, and the
entire file is linked below. We can see that these proteins show high similarity along their entire length (91% similarity over 1277 aa overlap). This indicates that there haven't been any major rearrangements, duplications, deletions or insertions, and likely no new domains since they diverged from a common ancestor. Even the N-terminal region is highly conserved, which is significant since N-termini of protein families often show tremendous polymorphism. |
 |
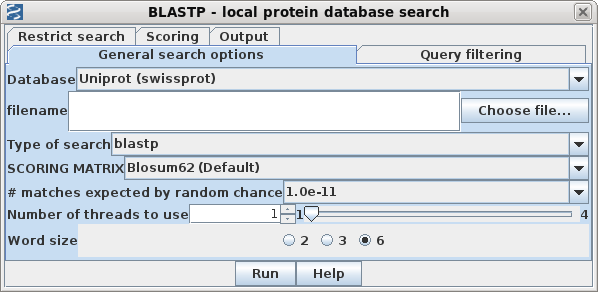
| In blprotein, select the 2020 sequence,
QHD43416, and choose Database --> BLASTP. (If you don't
have a local copy of Uniprot, use NCBI BLASTP instead.) Click Run to begin the search. Even on personal computers, results should pop up in less than a minute. |
 |
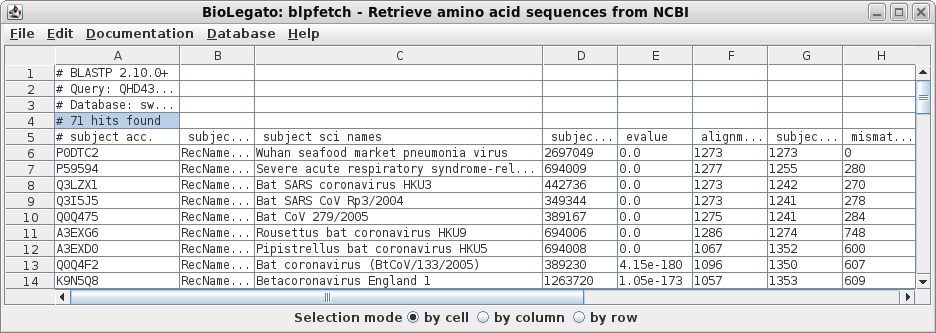
| Next, a tabular BLAST report will pop up in
blpfetch, a BioLegato application for viewing and retrieving
hits. |
 |
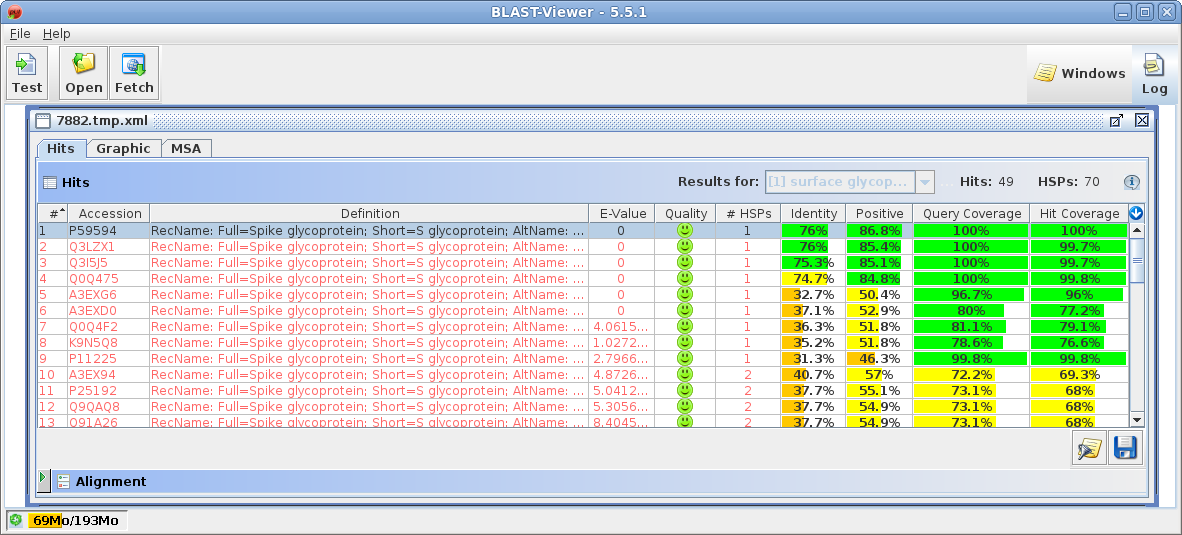
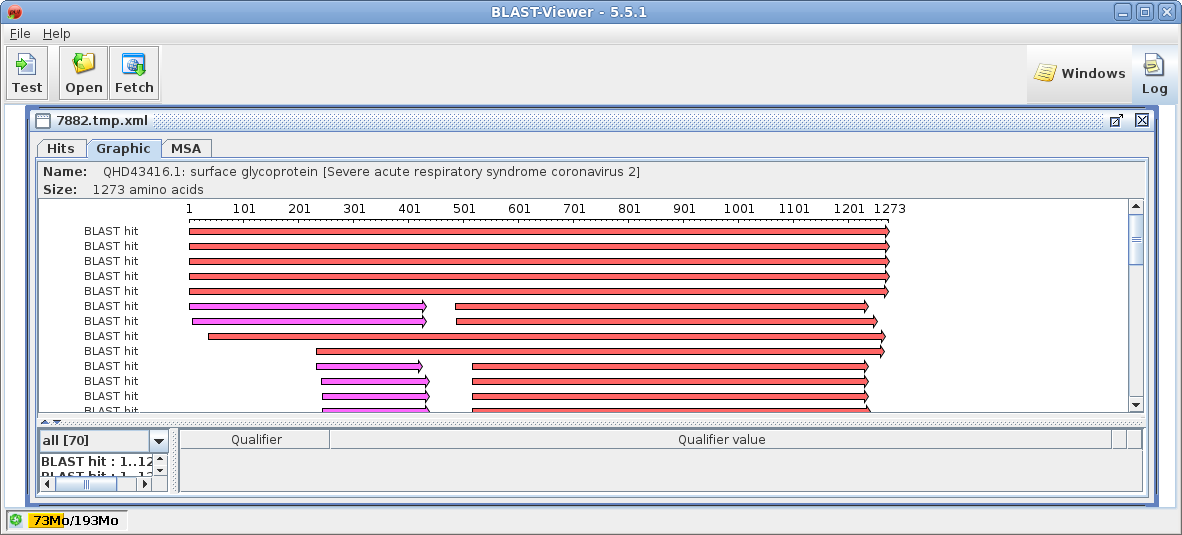
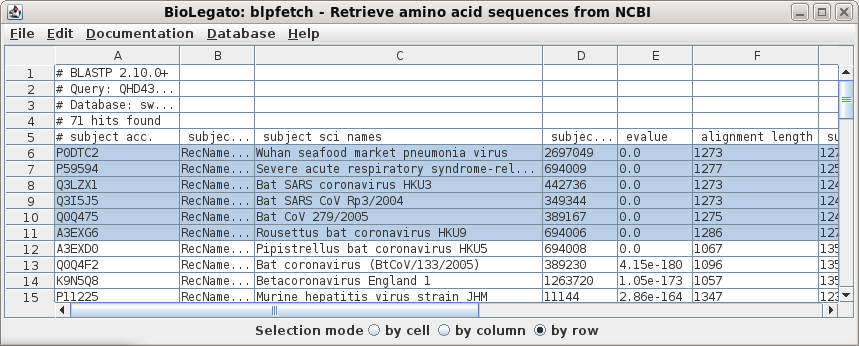
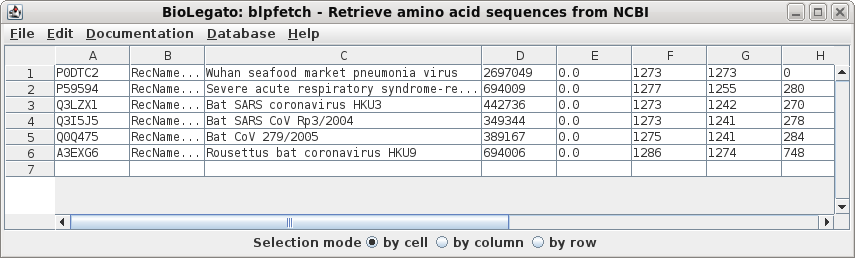
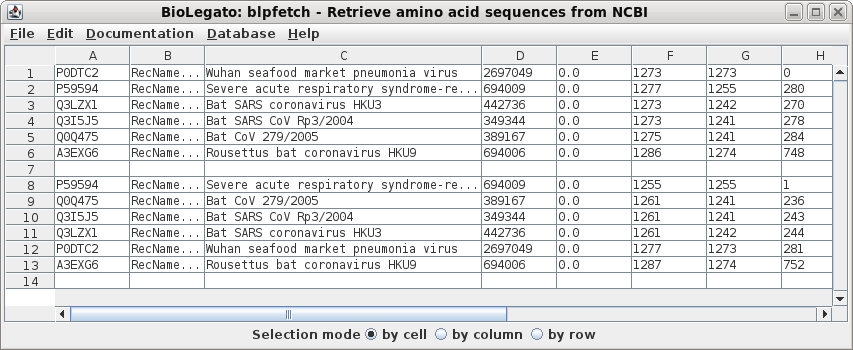
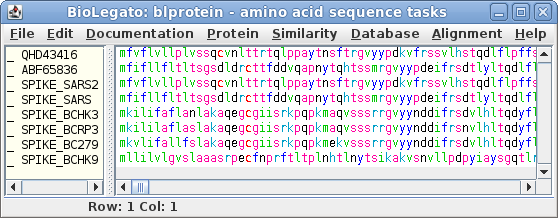
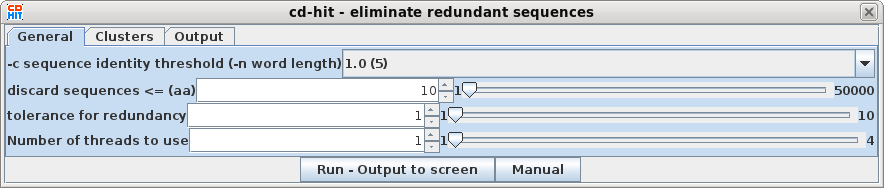
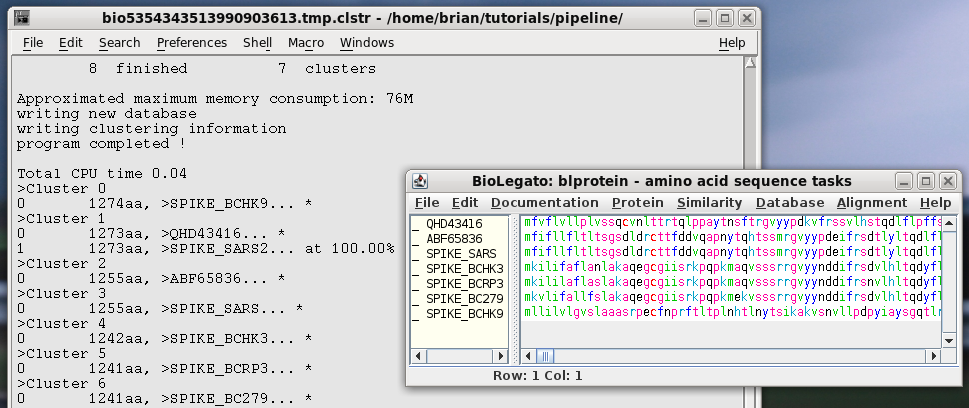
| Select all sequences in blprotein. Next choose Alignment --> cd-hit. Set the sequence identity threshold to 1.0, so that only perfectly matching sequences will be eliminated as duplicates. Click on Run |
 |
| Choose Alignment --> MAFFT. MAFFT has numerous
alignment algorithms based which vary the number of iterative
improvement to the alignment. As well, while most models consider the
entire alignment as a single conserved block, E-INS-i assumes multiple
conserved blocks punctuated by unalignable sites, and L-INS-i with 1
conserved block flanked by unalignable sites. By default, MAFFT will do a global alignment, and will choose the best algorithm based on the number of sequences. |
 |
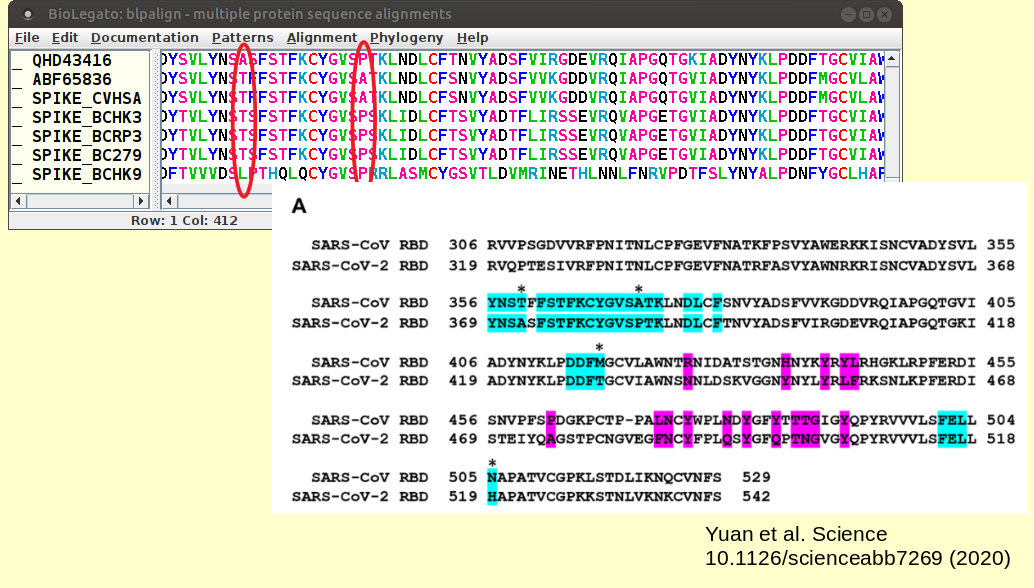
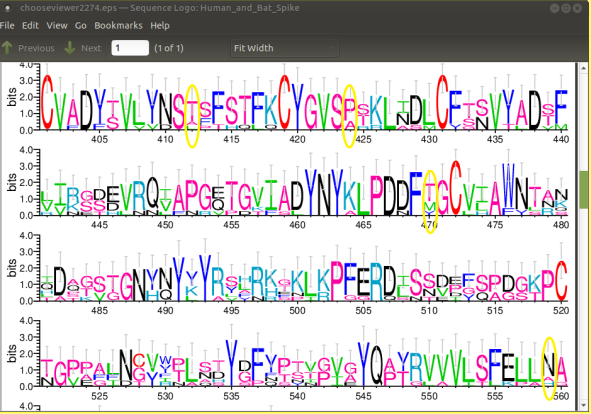
We may be able to learn more by looking at a sequence logo at these sites. Select all sequences in the alignment and choose Patterns --> Weblogo. |
 |
We can see replacements at the circled positions:
In all cases, the amino acid substitutions occur at sites of moderate information content, flanked on either side by sites with high information content. |
 |
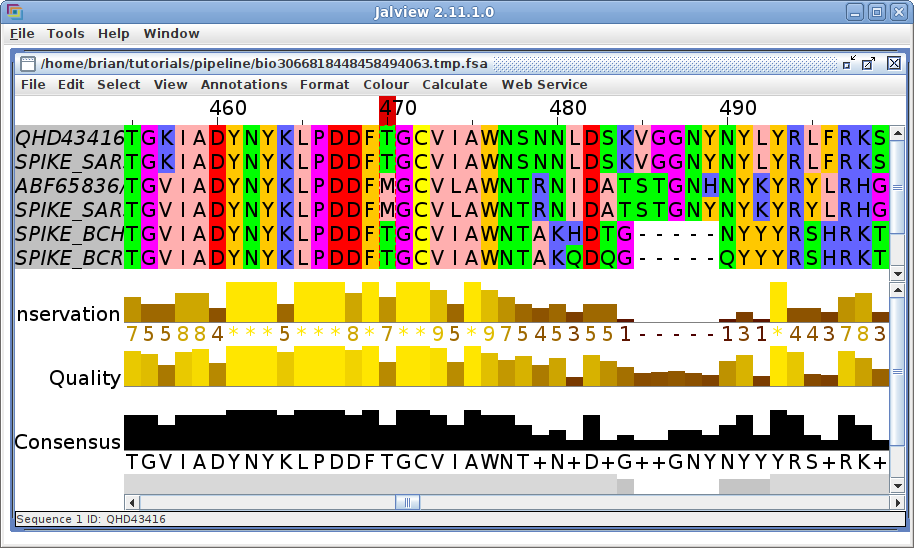
| Edit --> Select all Alignment --> Jalview Format --> Font=20 Color --> Zappo |
 |