Lecture 7, part 1 of 4
| PLNT4610/PLNT7690
Bioinformatics Lecture 7, part 1 of 4 |
Felsenstein J PHYLIP documentation [http://home.cc.umanitoba.ca/~psgendb/doc/Phylip/main.html]
Fitch WM and Margoliash E (1967) Construction of phylogenetic trees. Science 155:279-284.
Hershkovitz MA and Leipe DD (1998) Phylogenetic Analysis In Baxevanis AD and Ouellette BFF. Eds. Bioinformatics: A practical guide to the analysis of genes and proteins. John Wiley, Toronto.
Hillis DM, Allard MW, Miyamoto MM (1993) Analysis of DNA sequence data: Phylogenetic Inference. Meth. Enz. 224:456-487.
Setubal, J. and
Meidanis, J. (1997) Introduction to Computational Molecular
Biology. PWS Publishing Co., Toronto. Ch. 6 "Phylogenetic
Trees".
Tutorial: Maximum parsimony and maximum likelihood
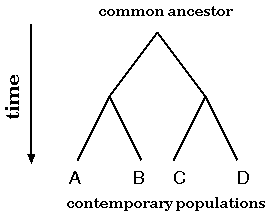
Evolution of genes or species can be modeled as a bifurcating tree
To a first approximation, the evolution of species or genes can be modeled as a birfurcating process. Two populations become reproductively isolated and diverge due to random mutational processes. Over time, this process may repeat itself, so that at any time, each population can be said to be most closely-related to some other population with which it shares a direct common ancestor.
Divergence
consists
of changes in characters, such as amino acids in a protein, or
nucleotides in DNA. The longer two populations remain
reproductively isolated, the more divergence will occur. In
principle, given the states for homologous characters across a
set of populations, it should be possible to work backwards in
time, ascending the tree, and reconstructing the character
states for each common ancestor in the tree, until a common
ancestor of all populations in the set is reached.
Reproductive isolationThe most important assumption in any phylogenetic model is that of reproductive isolation, such that no horizontal gene flow has occurred. However, speciation can often be a gradual process. Until reproductive isolation becomes complete, re-hybridization of closely-related species can result in horizontal gene flow. A good example is the recent evolutionary history of species related to wheat.As illustrated at right, modern wheat (Triticum aestivum) is a hexaploid containing three genomes, the A, B and D genome. Each genome is inferred to have arisen from the same common ancestor, but diverged into separate species. However, subsequent hybridization events have produced cereal species with all combinations of genomes, of which wheat is only one example. Horizontal gene flow is even more critical when considering prokaryotic genomes. Prokaryotes appear to have fewer fundamental barriers to horizontal gene transfer. Among prokaryotes, horizontal DNA transfer is relatively easy. At the other extreme, higher eukaryotes have germlines that are physically sequestered from contact with foreign species. |
 |
Heavily gapped regions are a major source of
error in multiple alignments
|
The alignment shows a region with gaps flanked by non-gapped sequence. This heavily gapped region illustrates two points. First, regions with large numbers of gaps are probably poorly aligned. For example, it might be more legitimate to align the block of A's at position 13 with the A's at position 10. However, that would require additional gaps, which cause a lower alignment score. Put another way, we are uncertain whether those A's in each case are homologous, that is similar due to common ancestry, or whether they represent independent positions in these proteins.
|
|
Therefore, where parts of an
alignment are uncertain due to gaps, it is often best to
delete those parts from the alignment before performing
phylogenetic analysis.
For those gaps that remain in the alignment, the problem of scoring arises. It is most likely that gaps spanning more than one position represent single insertion/deletion events. In that case, it is unreasonable to allow each gap position to contribute to the score during tree construction. One approach that is often used is to change all but the first gap in a long insertion/deletion region as unknowns. That is, most programs represent gaps as either dashes (-) or periods (.), and unknown positions as question marks (?). Thus programs that correct for incomplete data at some positions make it possible to work with a more realistic model of gaps.
| The figure shows a multigene family in some ancestral species, consisting of four copies of a gene. As speciation progresses, deletions and duplications occur independently in each species. In the lineage on the left, copies A and C are lost, and both B and D are duplicated. In the lineage leading to species 2, copy D is lost, and copies B and C are tandemly duplicated, followed by deletion of one copy of B. This model predicts that the longer since the divergence of two species, the fewer orthologous copies of a gene will remain, unless there is selective pressure to retain specific orthologous copies. However, while orthologous copies persist, they will cluster together on a tree. For example, B orthologues in species 1 will be more closely-related to B orthologoues in species2, than they will be to D orthologoues in species 1. Consequently, there is no guarantee that multiple copies of a gene will always be more closely-related to other copies within a species, than they will be to copies in other species. |
|
The more rapidly gene
families undergo duplication/deletion events, the more likely it
is that most or all copies of a gene within a species cluster
together on the same branch of a tree.
Finally, if gene
conversion results in the complete replacement of one copy of a
gene with another, it is functionally equivalent to a deletion
of one copy and duplication of another. However, it is possible
for partial gene conversion to result in replacement of only
part of a gene. In that case, a partially converted copy would
not be consistent with any simple model of evolution. That is,
two domains of a protein might have different evolutionary
histories. This phenomenon is referred to as 'reticulate
evolution'.
rRNA
DNA/RNA
protein
1homoplasy - return of a character to its
previous state, thus masking intervening mutational events.
Homoplasies are most important in DNA sequences, because there
are only 4 nucleotides. The magnitude of the problem: at any
position, one out of four mutations should result in a
homoplasy.
| PLNT4610/PLNT7690
Bioinformatics Lecture 7, part 1 of 4 |

