TUTORIAL: Genome AssemblyPreprocessing
of sequencing reads
|
November 4, 2021 |
TUTORIAL: Genome AssemblyPreprocessing
of sequencing reads
|
November 4, 2021 |
| raw read files (Illumina,
paired end) |
insert size
(nt) |
| DL300_S1_L001_R1_001_sample.fastq.gz DL300_S1_L001_R2_001_sample.fastq.gz |
300 |
| DL400_S2_L001_R1_001_sample.fastq.gz DL400_S2_L001_R2_001_sample.fastq.gz |
400 |
| DL700_S3_L001_R1_001_sample.fastq.gz DL700_S3_L001_R2_001_sample.fastq.gz |
700 |
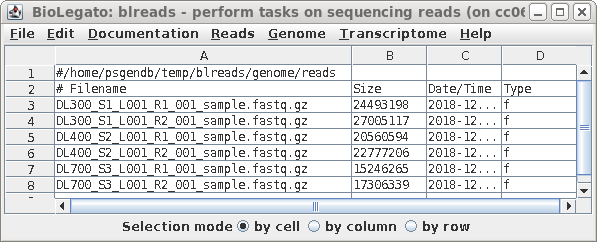
| Launch blreads by typing 'blreads &'.
(Here, we're adding & to the blreads command to run blreads in the background. That will allow us to continue using the command line in the same terminal window in which blreads was launched.) Note that the path for the current working directory is listed on the first line of blreads. Any line begining with a hash mark (#) is a comment, and will be ignored. |
 |
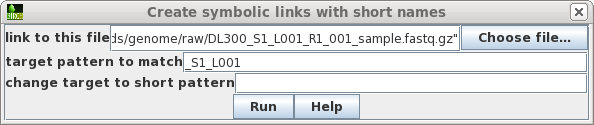
| Choose the first file to which you want to
make a link. To make the name of the link shorter, type in a target pattern that is common in two or more files. At right, the pattern is "_S1_L001". Since the short pattern field is left blank, this string will simply be omitted from the link name. The link name will be DL300_R1_001_sample.fastq.gz. |
 |
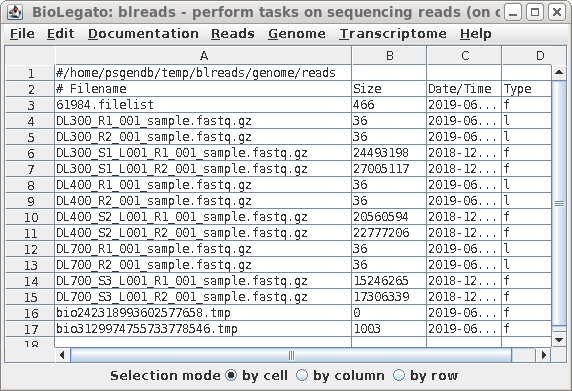
| The name of the link will appear in the
blreads window. Note that the type field of the original
file has 'f', to indicate a file, and the type field for the
link is 'l'. to indicate a link. |
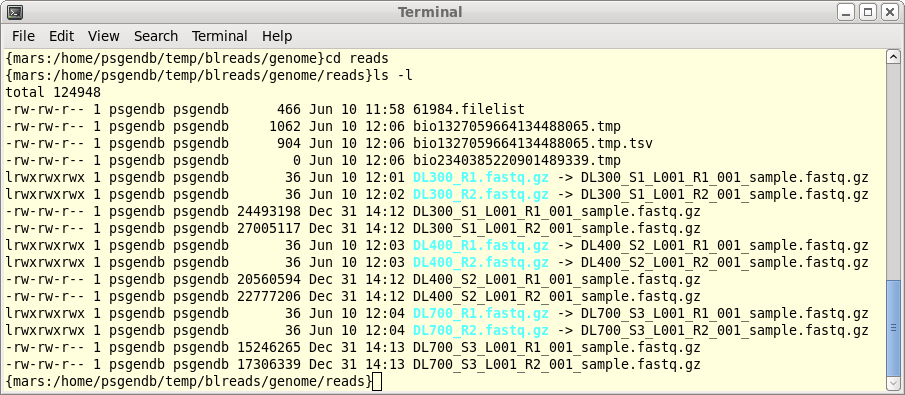
| When you have completed the process for all
six files, blreads should look like this: For each set of paired-end read files (R1 and R2), there will be a corresponding pair of symbolic links with shorter names. |
 |
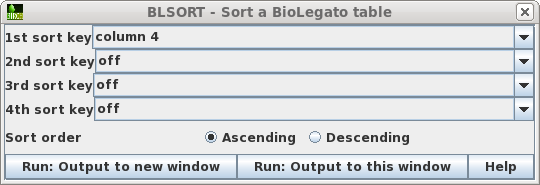
| To avoid accidentally changing the names of
the original files, first sort the files based on the Type
field, so that all links appear together in blreads. Choose
Edit --> BLSORT. Set the 1st sort key to column 4
(ie. Type), and Sort order to Ascending. Choose Run:Output
to this window. |
 |
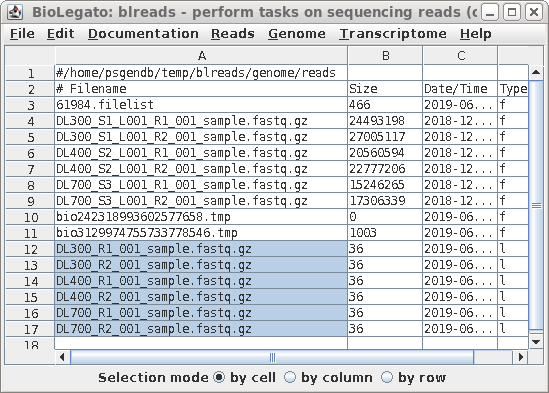
| All links will now be together in blreads.
Select the links as shown, and choose File --> Rename.
|
 |
| Set the target pattern to '_001_sample', and press Run. | 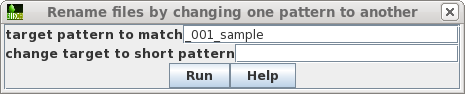 |
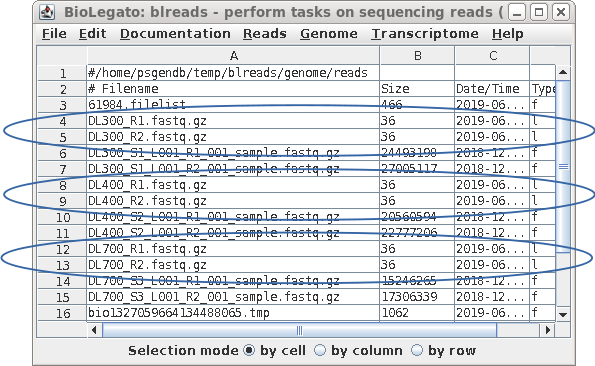
| Each pair of read files now has short, easy
to distinguish names, that will be used in all subsequent
steps. |
 |
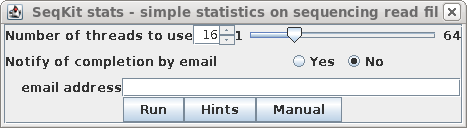
| By default, the number of threads used is 1/4
of the number of available cores, or 1, whichever is
greater. For most datasets, this is not a very time
consuming step. Therefore, it is usually unnecessary to use
additional cores. Simply click on Run. |
 |
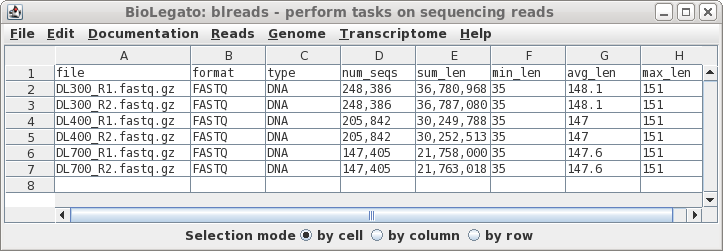
| In this case, all of the files look fairly
similar in terms of the number of reads and the average
length of reads. At this point, if there was a file that looked to be aberrant and you knew that it should be discarded, simply select the name of the bad file and delete the link using File --> Deletefiles. |
 |
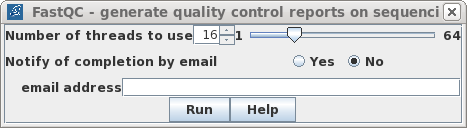
| Typical read files are larger than this
sample dataset, so with larger files, you may wish to speed
up FASTQC by setting a larger number of cores. In most
cases, results will appear soon enough that you don't need
to set notification of completion by email. Simply click on Run. |
 |
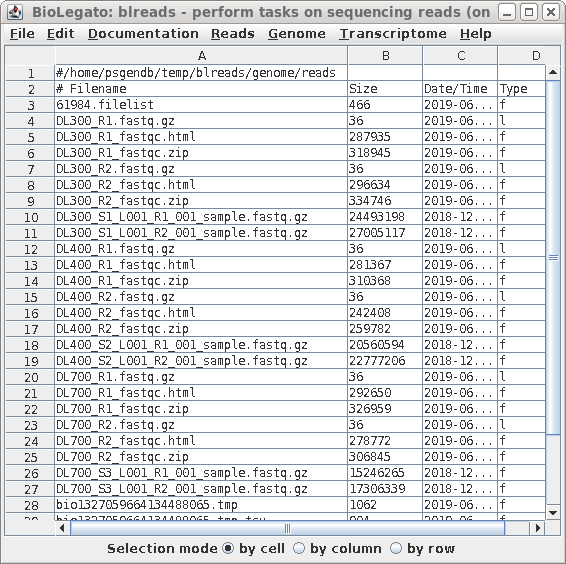
| blreads now lists the output files from
FASTQC. Files with the .html extension are viewable in any
web browser. The .zip files are the accompanying graphics
used by the web files. To view any report, select an HTML file and choose File --> View file. The HTML file will pop up in the browser. |
 |
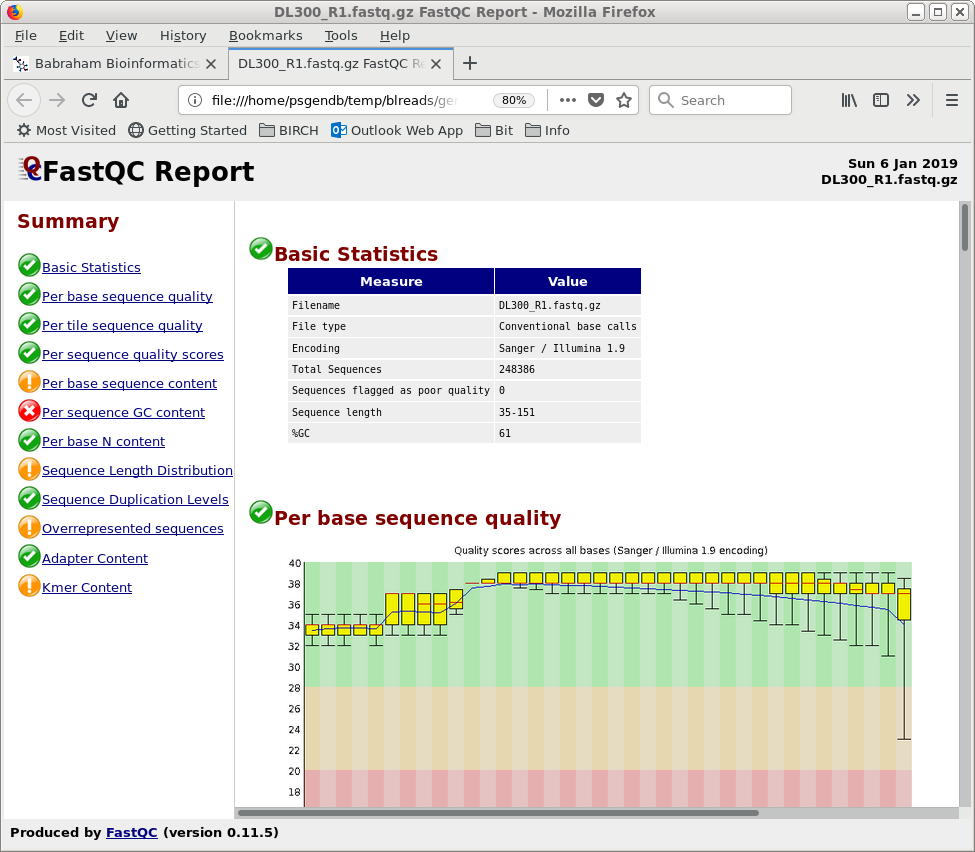
| For most datasets,
trim_galore does a good job of trimming adaptors off of
reads. However, for some paired-end datasets, trim_galore
may create 0-length reads, or eliminate a read altogether,
which confuses some assembly programs, particularly spades.
If such problems arise in downstream analysis, go back to
the this step and trim using Trimmomatic.
Trimmomatic is more complex to use, but results in
assemblies that are about the same as those using reads
trimmed by trim_galore. Trimmomatic can be run from blreads
using Reads --> Trimmomatic. |
| Most Illumina services use R1 and R2 as the
substrings which distinguish the left and right read pair
files for a given library, so by default, R1 and R2 is set
for "unique string for left/right reads" We only want to process read files, and not the other files in the directory. For this dataset, all read files use the .fastq.gz extension. Clicking on the Hints button will give a more detailed explantion of these parameters. |
 |
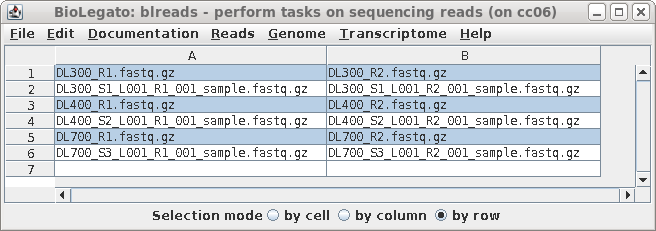
| A new blreads window will appear with the
left and right read files in two columns. Both the symbolic
links and the long-named read files appear. Since we prefer
to work with the short-named links, these can be selected by
clicking on Selection mode: row below, and then clicking on
the short-named read files. (This accomplishes the same
thing as sorting did above.) |
 |
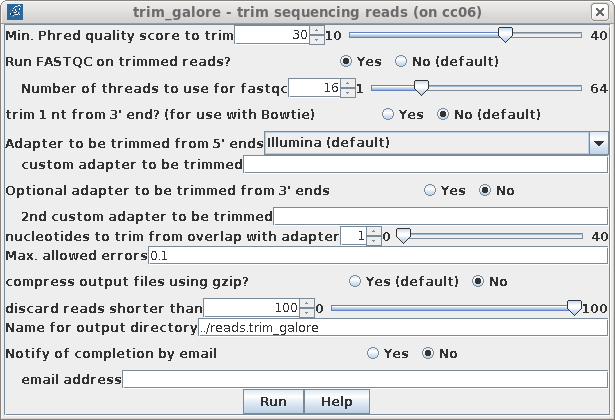
| While trim_galore has a lot of options, in
most cases you can run it with the defaults. One exception
would be to set the Min. Phred quality score to trim at 30,
rather than 20, which will shorten some reads, but the
remaining bases will be of higher quality. Based on the quality scores in the FASTQC files, we can also set discard reads shorter than to 100, because the number of reads in the Sequence Length Distribution reports show that the number of reads shorter than 100 are negligible. Set compress output to No, because some read correction programs such as Pollux can't work with compressed read files. By default, a new directory will be created for the output, specified in Name for output directory. The default will be to create a new directory called reads.trimmed in the parent directory. It is worth reading through the Trim_galore User's Guide before running trim_galore, especially if your reads use unusual adaptors. |
 |
| When trim_galore has processed all files, a
new blreads window will pop up. It is important to note that
this new window is running in the reads.trimmed directory.
At this point, one can probably close the previous blreads
window, which is running in the reads directory. The next
steps will be done in the reads.trimmed directory. First, notice that by default, trim_galore automatically runs FASTQC, so we have .zip and .html reports for each set of reads after trimming. For each read file, there are now three output files.
|
 |