|
Home
Contact
News
Research Group
Research Interests
General
Goals
Projects
Archive
Publications
Teaching
Open Positions
Undergraduate Projects
Department
University
Home
Contact
News
Research Group
Research Interests
General
Goals
Projects
Archive
Publications
Teaching
Open Positions
Undergraduate Projects
Department
University
Home
Contact
News
Research Group
Research Interests
General
Goals
Projects
Archive
Publications
Teaching
Open Positions
Undergraduate Projects
Department
University
Home
Contact
News
Research Group
Research Interests
General
Goals
Projects
Archive
Publications
Teaching
Open Positions
Undergraduate Projects
Department
University
|

General
Our research interests are in the areas of theoretical and computational chemistry, notably electronic structure theory. Computational quantum chemistry involves applying rigorous, 'first-principle' quantum-mechanical methods to the study of molecules, surfaces and solids and their properties. The growing relevance of quantum chemistry has been recognized, for instance, with the 1998 Nobel Prize in Chemistry that has been awarded to the late W. Kohn and J. Pople, two outstanding pioneers of the field.
More specifically, we are developing and applying state-of-the-art quantum chemical methods to study molecules, surfaces, interfaces, and solids and their properties. The specific quantum-chemical method that we use most often is known as density functional theory (DFT). Quantum-chemical simulations are computationally demanding, and powerful modern computer systems are required. (Here is a page describing our old group-level clusters.)
Computational quantum chemistry is uniquely positioned for the challenge of proposing and explaining chemically meaningful concepts and models such as, for instance, reaction mechanisms, bond orders, frontier orbitals or charges, their relationships to spectroscopic observables, and how and why these might change for a series of related compounds. This requires us to go beyond mere calculations, and focus on the interpretation of the calculated numbers also.
Many of our projects involve collaborations with other research groups − both experimental and theoretical − locally, across Canada and worldwide. If done well, collaborations enhance the research of all parties involved, and they provide unique opportunities for training and growth of students and postdocs.
Goals
The general, long-term goals of our research are as follows:
- To develop a solid understanding of the chemistry of actinides and other heavy elements from theory alone, so as to avoid difficult and dangerous experiments with radioactive and toxic materials (particularly transuranics) as much as possible; to facilitate their environmental remediation.
- To develop novel classes of high-performance materials for solar and other applications. Examples include the dye-sensitized solar cell (DSSC), singlet fission materials, conducting polymers and 2-dimensional (2D) materials.
- To contribute to a major increase in the role of solar energy, thus helping Canada respond to the global energy challenge and meet its obligations under COP21. Specifically, we aim at dramatic increases in the efficiency of solar cells.
- To develop computational chemistry, DFT methods in particular, into valuable and reliable research tools for areas where they have not yet reached their full potential, particularly regarding modeling complex systems (e.g. surfaces, interfaces, defects, heavy elements requiring relativity). This implies development of new and benchmarking and adaptation of existing methods.
|

| |
Research Projects and Themes
| Theoretical Actinide Molecular Science
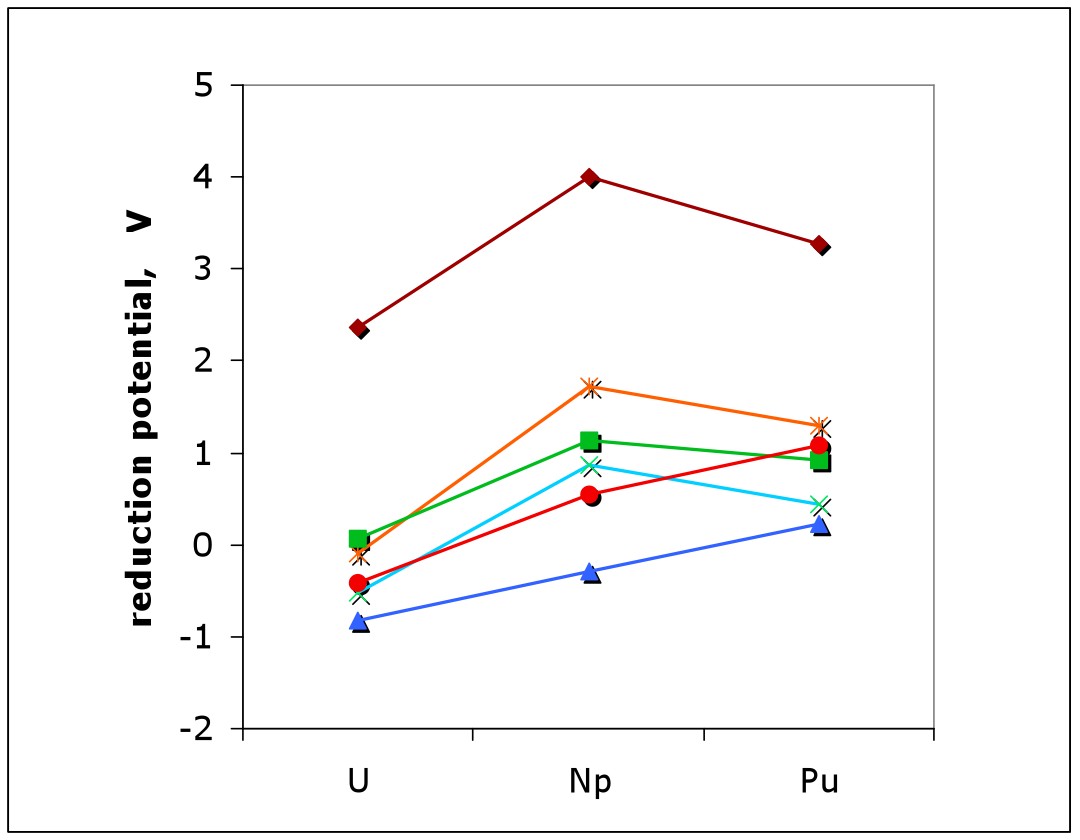
We use the tools of (relativistic) quantum chemistry to study the chemistry of the actinides, especially the early actinide elements (Th, U, Np, Pu). Recently, we have also extended this to the rest of the actinide series. Our research is motivated by fundamental interest arising from the unique chemistry of these elements, as well as by the nuclear waste problem. In many cases, it is also inspired by collaborations with experimental researchers.
Specific projects in the field are as follows:
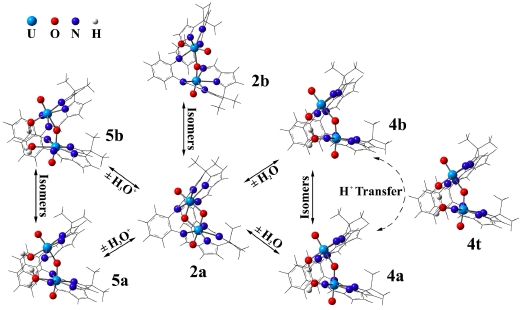
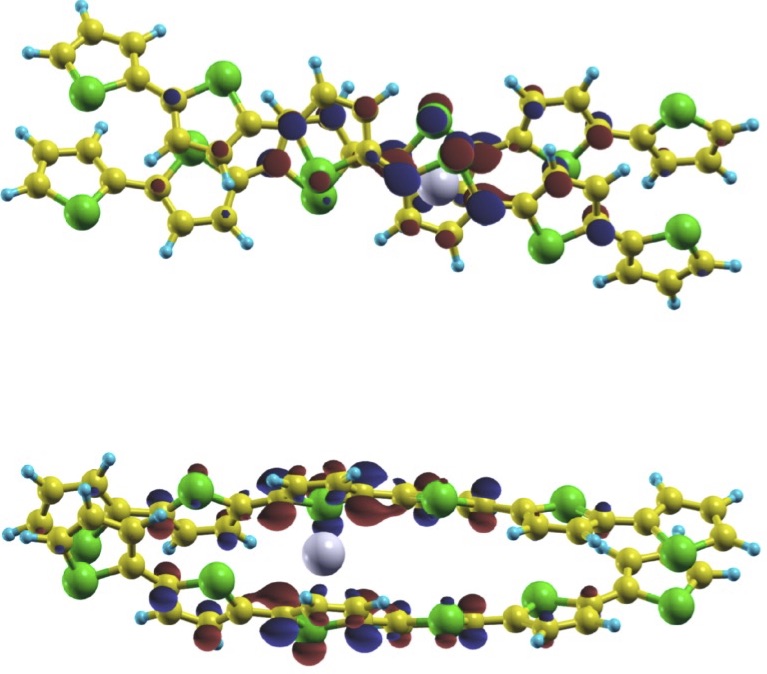
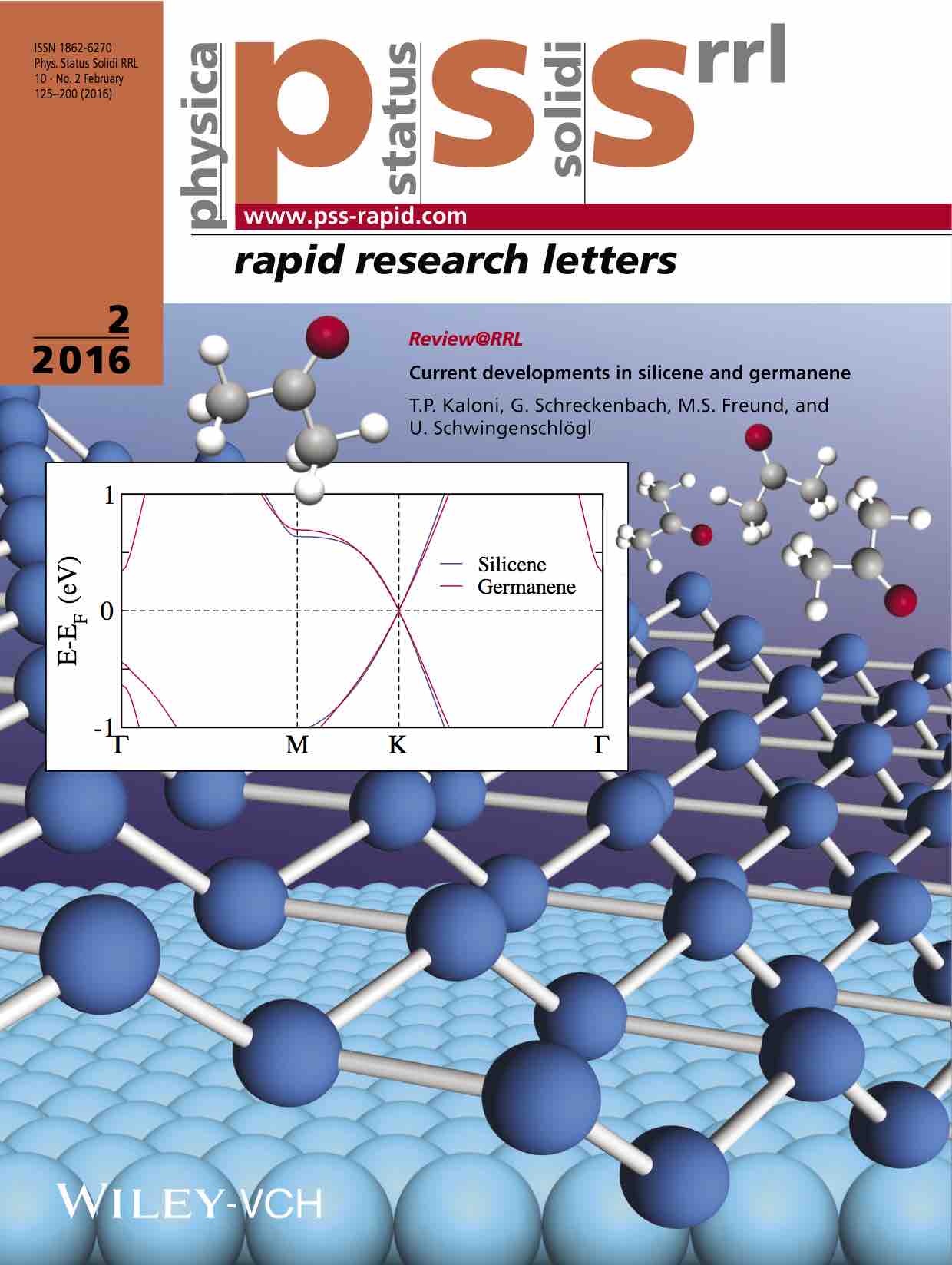
(i) Investigation of actinide inclusion complexes with polypyrrolic and other macrocycles, such as crown ethers or a "pacman"-like macrocycle that has two separate cavities. The "pacman" provides a platform for unique bond schemes involving actinide metals.
(ii) Aqueous (environmentally relevant) actinide species.
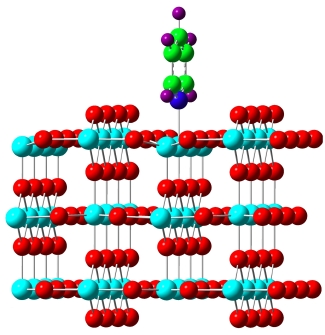
(iii) study of the interaction between aqueous actinide species and mineral surfaces, notably TiO2.
(iv) Interactions of actinides with 2D materials (vide infra).
(v) Periodic trends along the actinide series; complexes of later actinides (such as Cf and Es).
(vi) Chemistry of early actinides actinium, thorium and protactinium, for the purpose of proposing novel radiotherapeutics (Ac), or in comparison to uranium.
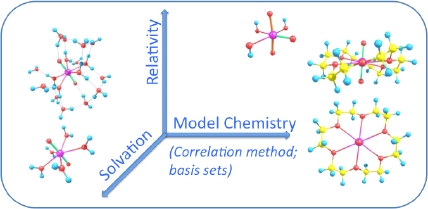
(vii) We are continuously involved in critically evaluating ("benchmarking") quantum-chemical methods as applied to the chemistry of the actinides. Three principal levels of approximation need to be optimized (relativistic approximation, model chemistry, solvation models).
We have reviewed our earlier work in computational/theoretical actinide chemistry [see Acc. Chem. Res. 43 (2010), 19−29].
| 
| Materials
Modeling materials requires building models of increasing complexity, in order to capture the essence of the problem of interest. This goes along with the need to apply a variety of theoretical and computational approaches. We use both, molecular (quantum-chemical) codes and periodic boundary condition DFT codes, combined with either atom-centered or plane-wave basis sets.
Current or recent projects in this area include:
(i) Conducting polymers. We model (semi-)conducting polymers like polythiophene and its derivatives, with a goal of supporting diverse applications such as in novel memory devices, other electronics, or solar energy conversion. Questions include the influence of chemical modifications and doping on the electronic structure (band structure) and the effects of stacking on the properties. Both, molecular (oligomer) and periodic models are used.
(ii) Two-dimensional (2D) materials. 2D materials are a novel and exciting class of materials with often unique properties; graphene is the prototypical example and best-studied system. We use the tools of computational chemistry and materials physics to study a wide variety of 2D materials. For those, we determine a range of interesting and relevant properties, and how these properties change upon changing the material (e.g. along the series graphene, silicene, germanene) or through chemical modifications. This has been extended toward semiempirical tight-binding modeling of nanoribbons, nanoparticles and similar systems.
(iii) Materials for solar energy conversion are discussed separately below.
(iv) We develop novel computational tools, specifically with materials applications and the required complexity in mind; see below.
| 
| Solar Energy
'Energy' − extraction of fossil and nuclear fuels and the production of electricity (or transportation fuels) from these sources − is the single largest global industry. Access to cheap and plentiful energy, 'powering the planet', underpins much of modern society and lifestyle. Challenges related to energy are correspondingly huge, and many of these are related to the waste produced along the way. Indeed, global warming caused by human activity (primarily carbon dioxide emissions, the waste from burning fossil fuels) has emerged as perhaps the most pressing environmental challenge of our time. Reducing CO2 emissions requires invention, development and deployment of carbon-neutral energy production including solar energy conversion on a large scale.
Currently or recently, we have pursued a number of projects that apply the tools of computational chemistry to understanding molecular processes related to solar energy, and thus to contribute to the development of CO2-free energy sources:
(i) Dye-sensitized solar cell (DSSC). We study molecular-level processes within the DSSC, e.g. dye regeneration mechanism, or properties of dyes in relation to cell efficiency.
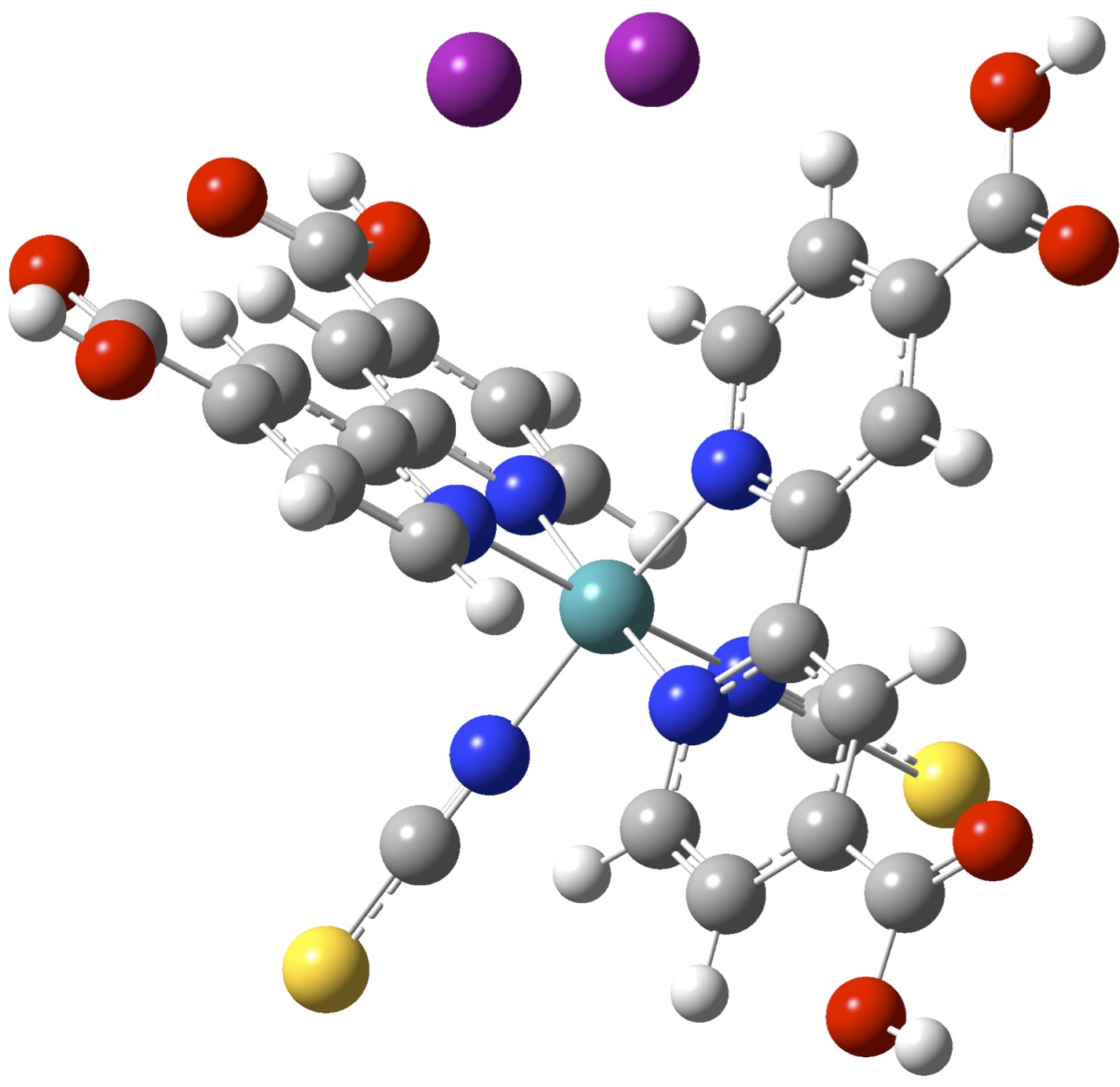
(ii) Computational screening of singlet fission materials.
(iii) Polymer-based photovoltaics.
(iv) Catalytic water splitting by Ru and U complexes. (Noting the intermittency of sunlight, an efficient way of storing energy − e.g. as chemical fuel − is crucial to the large-scale application of solar energy conversion.)
| 
| Method Development and Evaluation
We are working on the development of new quantum-chemical methods and on the evaluation ("benchmarking") of existing approximations, as applied to various chemical problems.
Currently or recently, this has included:
(i) Evaluation − benchmarking − of approximate DFT methods, relativistic methods, and continuum solvation models for applications to the chemistry of heavy elements.
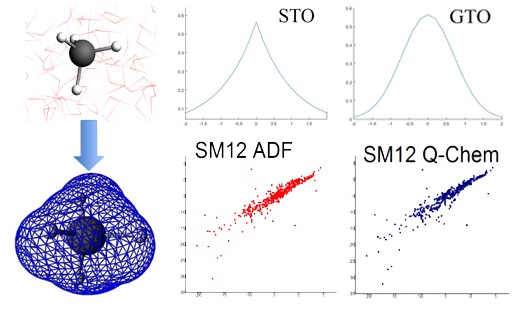
(ii) Development and implementation of continuum solvation models (SM12) for molecular and periodic calculations. This has been done within the environment of the ADF suite of programs.
(iii) Development of high-throughput computational (HTC) tools for the prescreening of DSSC dyes.
(iv) More realistic surface and interface models that include, for instance, defects or edges.
| 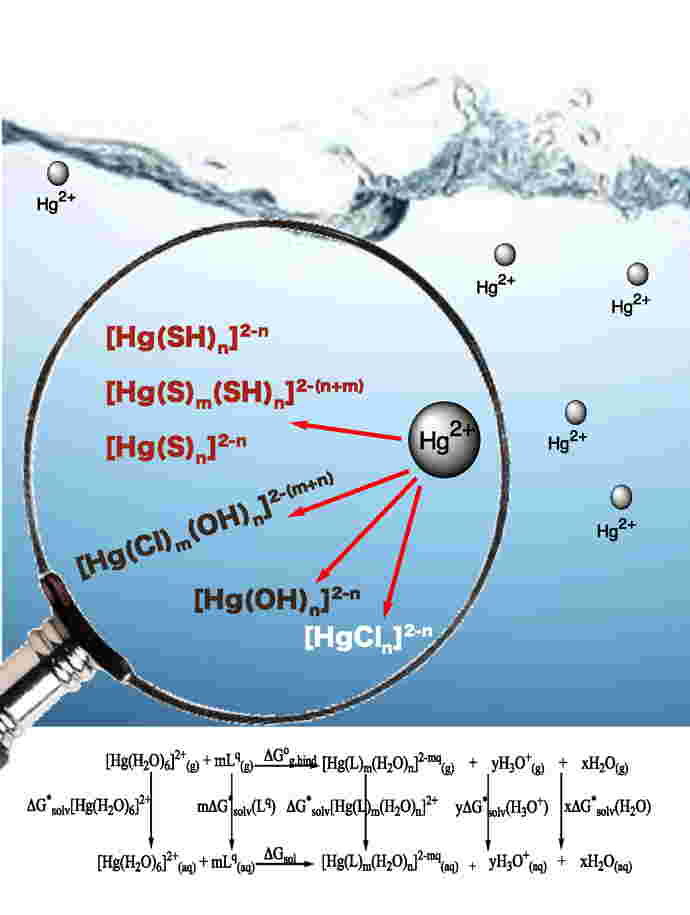
| Environmental Chemistry in Silico
We study molecular-level processes involving crude oil, mercury and other heavy metals, aiming at the environmental chemistry of these elements. This is often done in collaboration with environmental chemists.
Specifically, we have looked at the following aspects of environmental mercury chemistry:
(i) Novel fluorescence sensors for mercury;
(ii) Aqueous mercury species: structures and thermodynamic parameters;
(iii) Interactions of environmental mercury and other metals with ice surfaces;
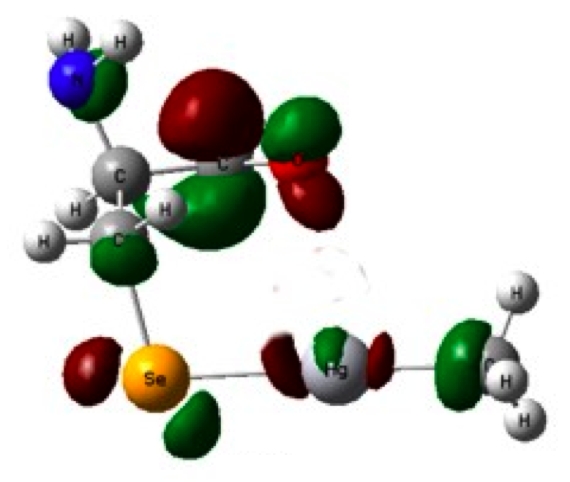
(iv) Molecular-level mechanism of the toxicological Hg−Se antagonism.
(v) Gas-phase reactions of Hg will be modeled in order to obtain accurate thermodynamic and kinetic parameters for reactions relevant to atmospheric mercury transport.
In addition, (vi), we are modelling thermodynamic parameters of crude oil components at cold temperatures (simulating an Arctic environment), for the purpose of supporting mitigation efforts for oil spills in the Arctic.
Parts of the actinide research theme (described above) fit here also (e.g. aqueous complexes; mineral surface interactions).
We have reviewed our work on environmental mercury chemistry [see Acc. Chem. Res. 52 (2019), 379−388].
|
|
