TUTORIAL:
Transcriptome
Assembly
|
June 18, 2019 |
TUTORIAL:
Transcriptome
Assembly
|
June 18, 2019 |
| Choose File --> guesspairs. As before, we use R1 and R2 to identify paired-end read files, and ask blreads to only select files with the .fq file extension. |
 |
| A new blreads window appears with the
paired-end files in two columns. Select all pairs of reads,
and choose Transcriptome --> rnaspades. |
 |
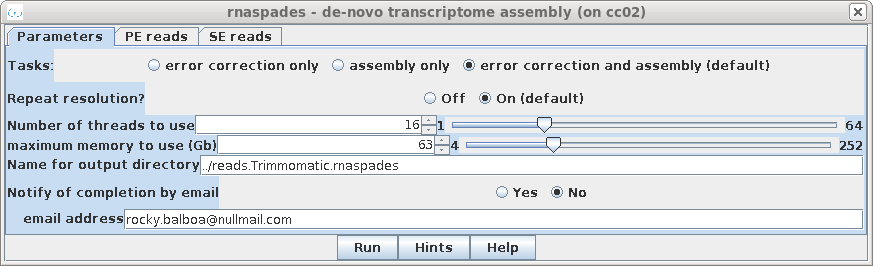
| Set the output directory
../reads.Trimmomatic.rnaspades. Since it takes a long time to do an assembly, it is best to set Notify of completion by email to Yes, and type in your email address. If you had additional files with single-end reads reads or additional paired-end reads those files can be selected in the SE reads or PE reads (not shown) tabs, respectively. |
  |
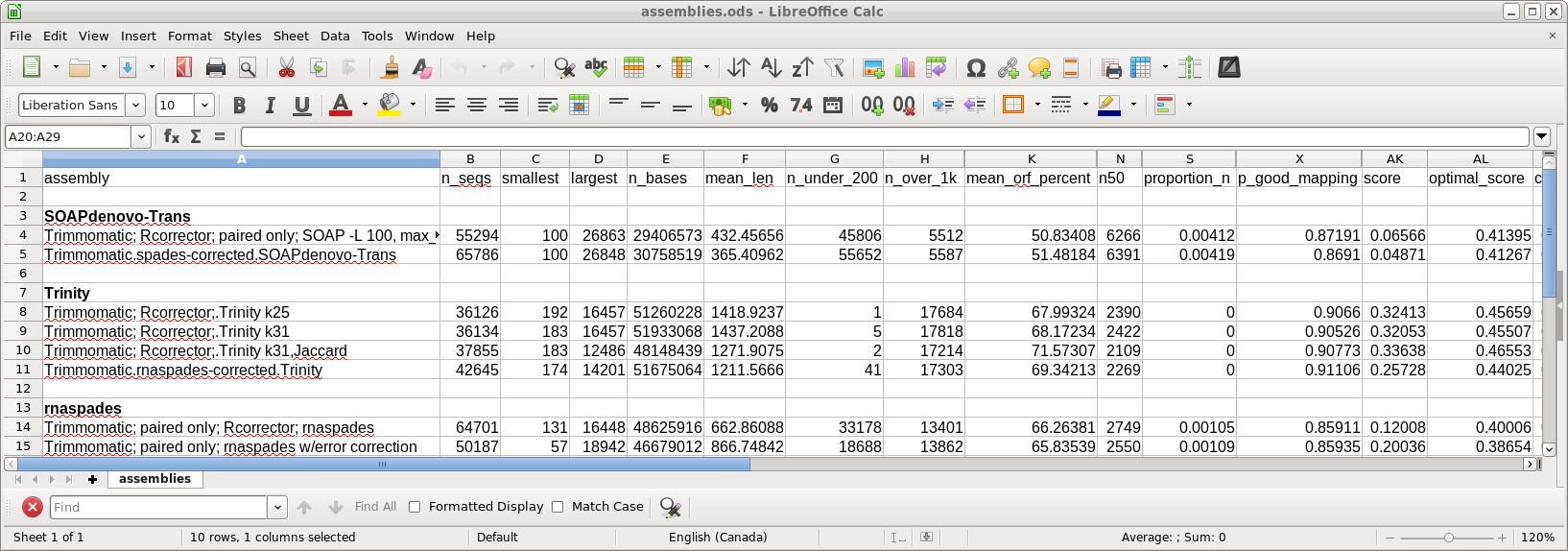
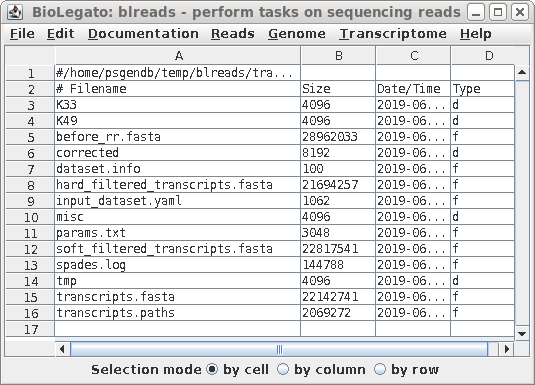
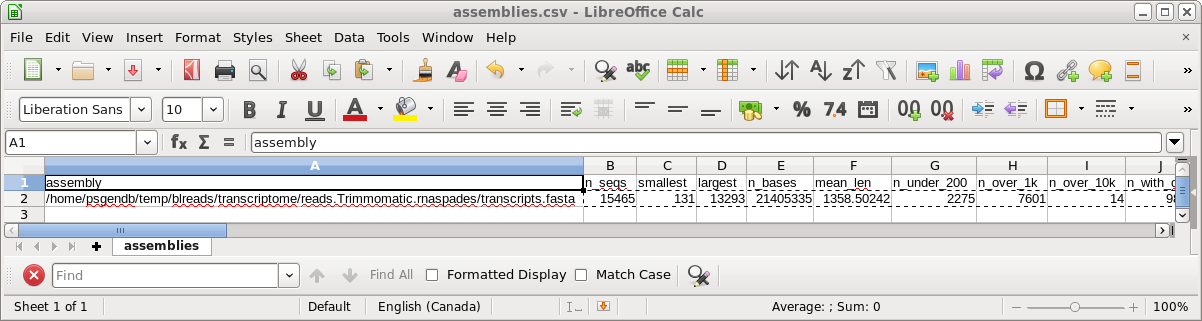
| The final contigs will be found in
contigs.fasta, and scaffolds in transcripts.fasta. To see a summary of the results, select spades.log and choose File --> View file. |
 |
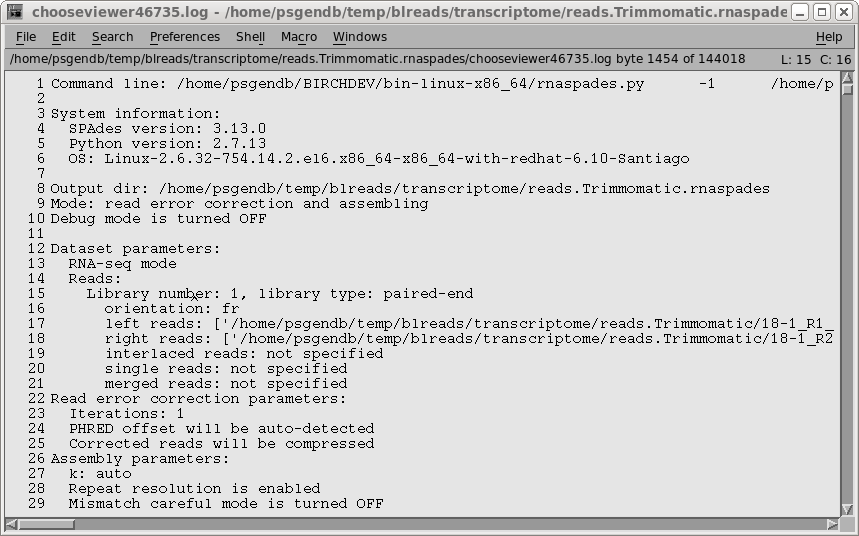
| The parameters of the assembly, along with a
progress report, are shown spades.log. These results don't tell us much about the assembly, so in the next step we will run transrate to evaluate the assembly. |
 |
| The starting point for transrate is to go
back to the same blreads window that has the read pairs (in
the reads.Trimmomatic directory and again select the read
pairs. (If you closed that window, you will need to open
blreads in the reads.Trimmomatic directory, and run
guesspairs as before. Next, choose Transcriptome --> transrate. The transcriptome assembly file, transcripts.fasta, is in the rnaspades directory from the last step. By default, output is written to a directory called transrate. |
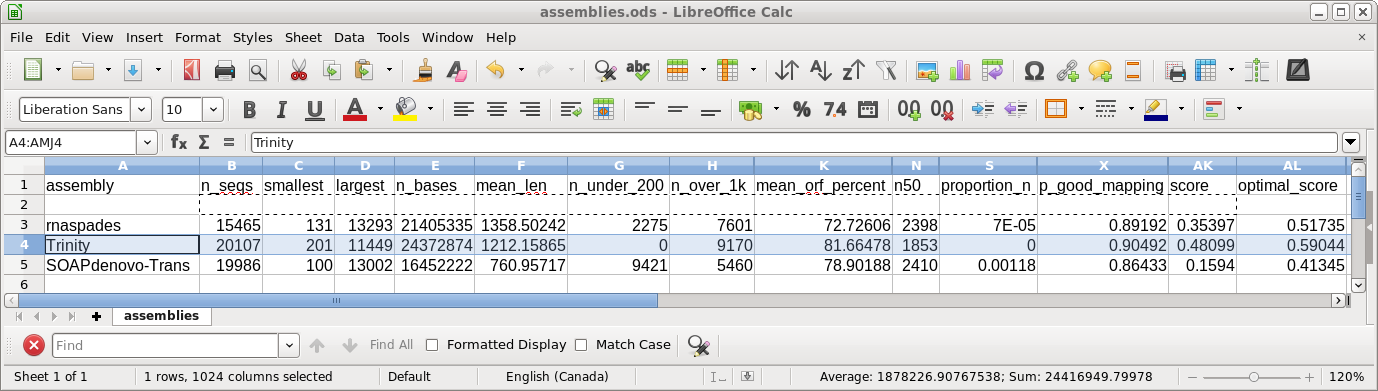
| transrate.log - the details of the
transrate run. The most important parameter is the TRANSRATE ASSEMBLY SCORE, which for this assembly is 0.35. While the published scores tend to be closer to the OPTIMAL scores, remember that the dataset for this tutorial has only 5% of the total reads, meaning that coverage is less. Assemblies done using higher coverage will give higher scores. The complete output can be seen in transrate.log. |
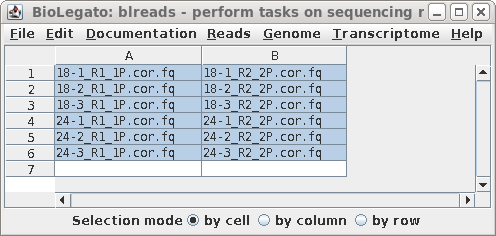
| For the assemblies using Trinity and
SOAPdenovo-Trans, we will use the read files corrected by
Rcorrector in the previous section. Open blreads in reads.Trimmomatic.Rcorrector and run guessreads to get all files with the .fq file extension. Select all read pairs. |
 |
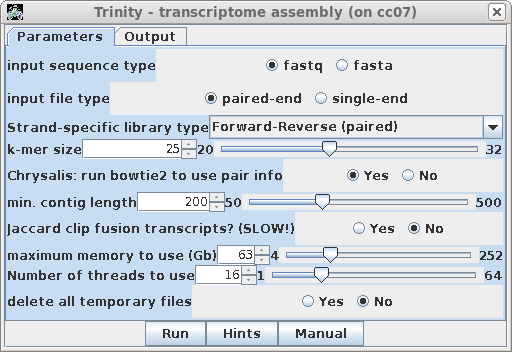
| For this dataset there is probably no need to
change the parameters. It may be worth trying several k-mer sizes, but keep in mind that for RNAseq, where reads are usually 100 nt, larger k-mers will artificially lower the coverage. If you wanted to make sure to assemble tRNAs or other small RNAs in the assembly, set min. contig length to the size of the RNA you want to assemble. In the Output tab, set output directory to ../reads.trim_galore.Rcorrector.Trinity. |
 |
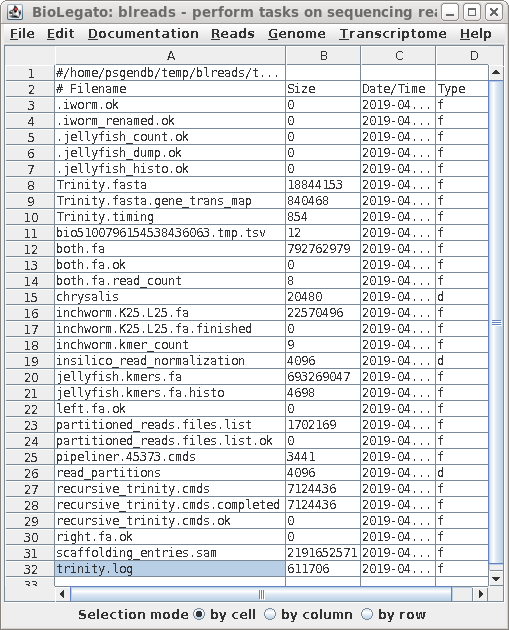
| A progress report of the assembly can be
found in the file trinity.log. Other than verifying that no
errors occurred in the assembly, this file doesn't provide a
lot of information on the assembly itself. The final set of transcripts is found in Trinity.fasta. Genes vs. Transcripts - Remember: In eukaryotes, any gene may potentially produce 2 or more different transcripts as a result of either alternative splicing of introns, or alternative transcription start sites. Consequently, the number of transcripts in a transcriptome will be greater than the number of genes in the genome. |
 |
| As done for the rnaspades output, run
Transrate to evaluate the results. Select the same read files used in the assembly and choose Transcriptome --> transrate. Select Trinity.fasta as the assembly file. The TRANSRATE ASSEMBLY SCORE of 0.481 is a substantial improvement over the rnaspades assembly. The p good contigs score of 0.94 is about as high as you ever see for a transcriptome assembly. |
| As with Trinity, set shortest contig to 200,
keeping in mind that smaller settings are needed if you want
to assemble small RNAs. (Although you can run SOAPdenovo-Trans with k-mers up to 127, it seldom makes sense to do so, since for RNAseq reads are necessarily short eg. 100 nt.) |
| In the Output tab, set the output directory
to ../reads.Trimmomatic.Rcorrector.SOAPdenovo-Trans. SOAPdenovo-Trans lets you specify a prefix to be used for all output filenames, so in this case we'll set the prefix to "Rdio" for R. diobovatum. |
| Next, using the same read files as before,
run Transcriptome --> transrate, using
Rdio.scafSeq as the assembly file. |
| Output from transrate.log is shown at right. The TRANSRATE ASSEMBLY SCORE is only 0.16, suggesting that SOAPdenovo-Trans doesn't work as well with low-coverage samples. |