TUTORIAL:
Transcriptome
Assembly
|
June 17, 2019 |
TUTORIAL:
Transcriptome
Assembly
|
June 17, 2019 |
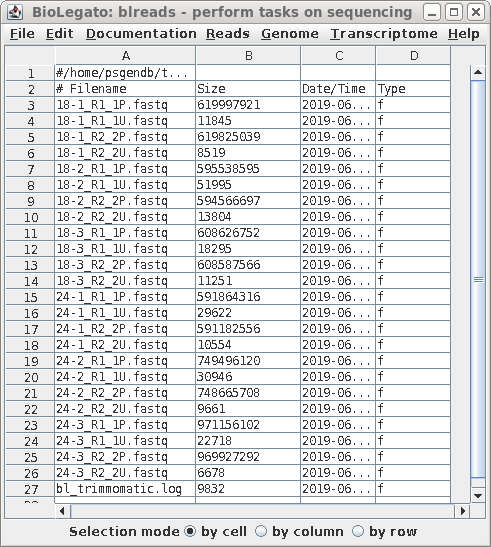
| Output from Trimmomatic will have the .fq
file extension, so choose Yes for Only process...
and enter the P.fastq file extension. This will
select only the paired reads, ignoring the unpaired reads. |
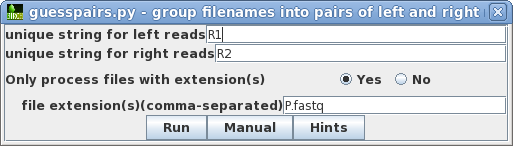 |
| Clicking on Run will bring up a new
blreads window with the best quess of file pairing, in two
columns. Usually, guesspairs.py gets it right. Files for
which a pair cannot be found (ie. single-end reads) would be
listed in a single column. To run Rcorrector with these read pairs, choose Edit --> SelectAll, and then Reads --> Rcorrector. |
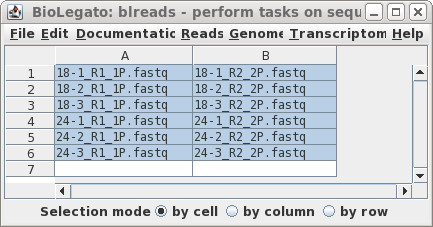 |
| By default, Rcorrector will write corrected
reads to a directory in the parent directory (ie. not within
reads.trimmed) called reads.trimmed.Rcorrector. Let's change
that to reads.Trimmomatic.Rcorrector. Rcorrector may take awhile to run, so you may wish to set "Notify of completion by email" to Yes and type in an email address. (On this dataset, Rcorrector took about 8 min. to complete using 16 threads.) |
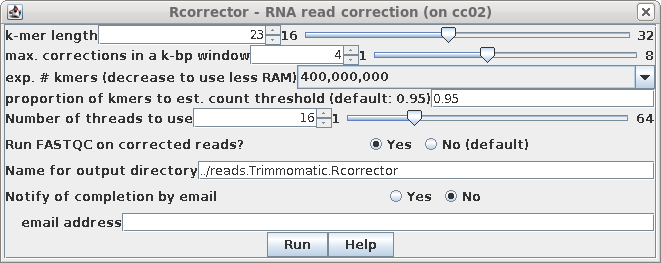 |
| The output is written to the
reads.Trimmomatic.Rcorrector directory, whose contents is
shown at right. |
 |