TUTORIAL: Transcriptome AssemblyPreprocessing
of RNA sequencing reads
|
October 31, 2021 |
TUTORIAL: Transcriptome AssemblyPreprocessing
of RNA sequencing reads
|
October 31, 2021 |
| raw read files (Illumina,
paired end) |
time |
replicate |
| HI.3992.004.Index_1.18-1_R1_S5.fastq.gz HI.3992.004.Index_1.18-1_R2_S5.fastq.gz |
18 h |
1 |
| HI.3992.004.Index_3.18-2_R1_S5.fastq.gz HI.3992.004.Index_3.18-2_R2_S5.fastq.gz |
18 h |
2 |
| HI.3992.004.D701---D503.18-3_R1_S5.fastq.gz HI.3992.004.D701---D503.18-3_R2_S5.fastq.gz |
18 h |
3 |
| HI.3992.004.Index_8.24-1_R1_S5.fastq.gz HI.3992.004.Index_8.24-1_R2_S5.fastq.gz |
24 h |
1 |
| HI.3992.004.Index_10.24-2_R1_S5.fastq.gz HI.3992.004.Index_10.24-2_R2_S5.fastq.gz |
24 h |
2 |
| HI.3992.004.Index_11.24-3_R1_S5.fastq.gz HI.3992.004.Index_11.24-3_R2_S5.fastq.gz |
24 h |
3 |
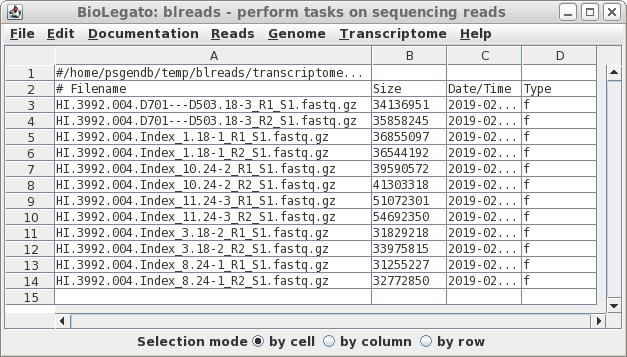
| Launch blreads by typing 'blreads &'.
(Here, we're adding & to the blreads command to run blreads in the background. That will allow us to continue using the command line in the same terminal window in which blreads was launched.) Note that the path for the current working directory is listed on the first line of blreads. Any line begining with a hash mark (#) is a comment, and will be ignored. |
 |
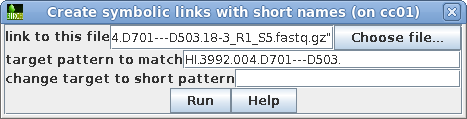
| Choose the first file to which you want to
make a link. To make the name of the link shorter, type in a target pattern that is common in two or more files. At right, the pattern is "HI.3992.004.D701---D503.". Since the short pattern field is left blank, this string will simply be omitted from the link name. The link name will be 18-3_R1_S5.fastq.gz. |
 |
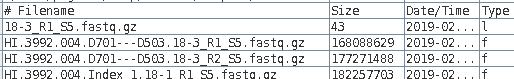
| The name of the link will appear in the
blreads window. Note that the type field of the original
file has 'f', to indicate a file, and the type field for the
link is 'l'. to indicate a link. |
 |
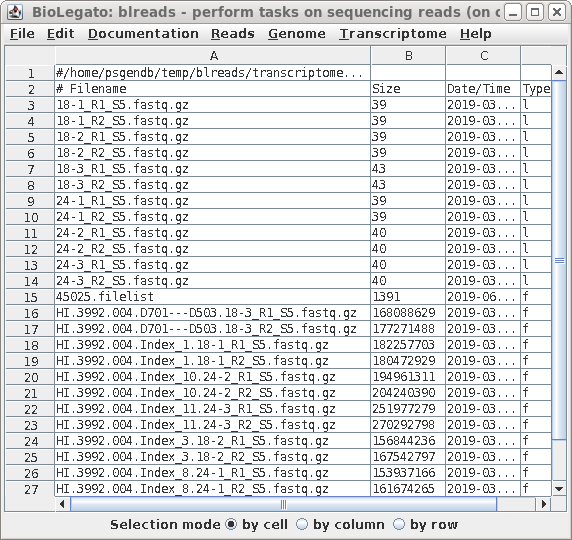
| When you have completed the process for all
six files, blreads should look like this: For each set of paired-end read files (R1 and R2), there will be a corresponding pair of symbolic links with shorter names. |
 |
| lrwxrwxrwx 1 psgendb
psgendb 39 Feb
26 12:26 18-1_R1_S5.fastq.gz ->
HI.3992.004.Index_1.18-1_R1_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 39 Feb 26 12:26 18-1_R2_S5.fastq.gz -> HI.3992.004.Index_1.18-1_R2_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 39 Feb 26 12:26 18-2_R1_S5.fastq.gz -> HI.3992.004.Index_3.18-2_R1_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 39 Feb 26 12:26 18-2_R2_S5.fastq.gz -> HI.3992.004.Index_3.18-2_R2_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 43 Feb 26 12:26 18-3_R1_S5.fastq.gz -> HI.3992.004.D701---D503.18-3_R1_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 43 Feb 26 12:26 18-3_R2_S5.fastq.gz -> HI.3992.004.D701---D503.18-3_R2_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 39 Feb 26 12:26 24-1_R1_S5.fastq.gz -> HI.3992.004.Index_8.24-1_R1_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 39 Feb 26 12:26 24-1_R2_S5.fastq.gz -> HI.3992.004.Index_8.24-1_R2_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 40 Feb 26 12:26 24-2_R1_S5.fastq.gz -> HI.3992.004.Index_10.24-2_R1_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 40 Feb 26 12:26 24-2_R2_S5.fastq.gz -> HI.3992.004.Index_10.24-2_R2_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 40 Feb 26 12:26 24-3_R1_S5.fastq.gz -> HI.3992.004.Index_11.24-3_R1_S5.fastq.gz lrwxrwxrwx 1 psgendb psgendb 40 Feb 26 12:26 24-3_R2_S5.fastq.gz -> HI.3992.004.Index_11.24-3_R2_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 20 Feb 26 12:25 bio803081030103253751.tmp.tsv -rw-rw-r-- 1 psgendb psgendb 168088629 Feb 26 11:54 HI.3992.004.D701---D503.18-3_R1_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 177271488 Feb 26 11:54 HI.3992.004.D701---D503.18-3_R2_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 194961311 Feb 26 07:57 HI.3992.004.Index_10.24-2_R1_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 204240390 Feb 26 07:57 HI.3992.004.Index_10.24-2_R2_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 251977279 Feb 26 07:57 HI.3992.004.Index_11.24-3_R1_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 270292798 Feb 26 07:57 HI.3992.004.Index_11.24-3_R2_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 182257703 Feb 26 11:54 HI.3992.004.Index_1.18-1_R1_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 180472929 Feb 26 11:54 HI.3992.004.Index_1.18-1_R2_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 156844236 Feb 26 11:54 HI.3992.004.Index_3.18-2_R1_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 167542797 Feb 26 11:54 HI.3992.004.Index_3.18-2_R2_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 153937166 Feb 26 07:58 HI.3992.004.Index_8.24-1_R1_S5.fastq.gz -rw-rw-r-- 1 psgendb psgendb 161674265 Feb 26 07:58 HI.3992.004.Index_8.24-1_R2_S5.fastq.gz |
| Select the links as shown, and choose File
--> Rename. |
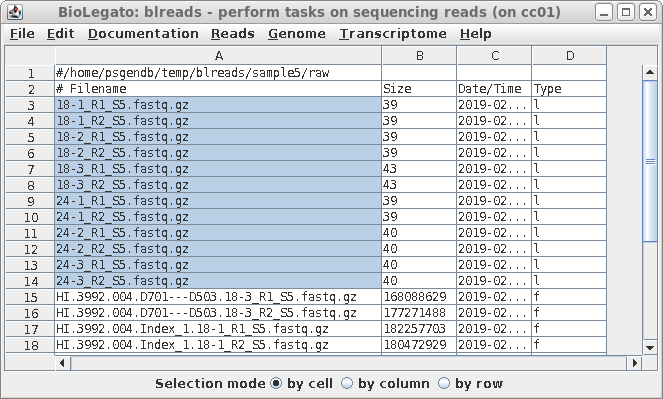 |
| Set the target pattern to '_S5', and press Run. | 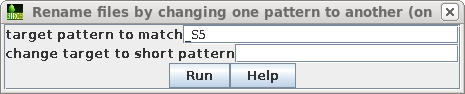 |
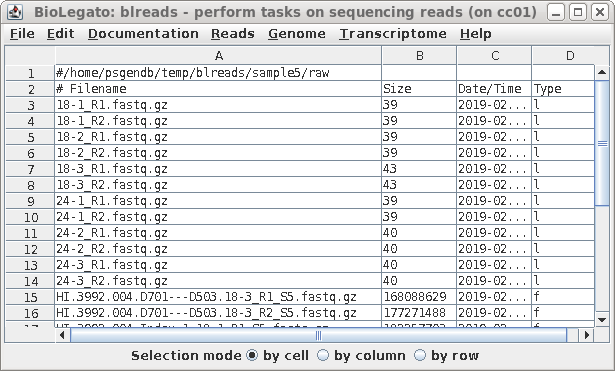
| Each pair of read files now has short, easy
to distinguish names, that will be used in all subsequent
steps. |
 |
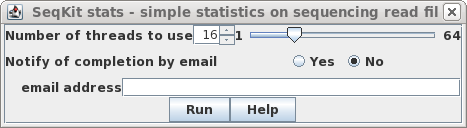
| By default, the number of threads used is 1/4
of the number of available cores, or 1, whichever is
greater. For most datasets, this is not a very time
consuming step. Therefore, it is usually unnecessary to use
additional cores. Simply click on Run. |
 |
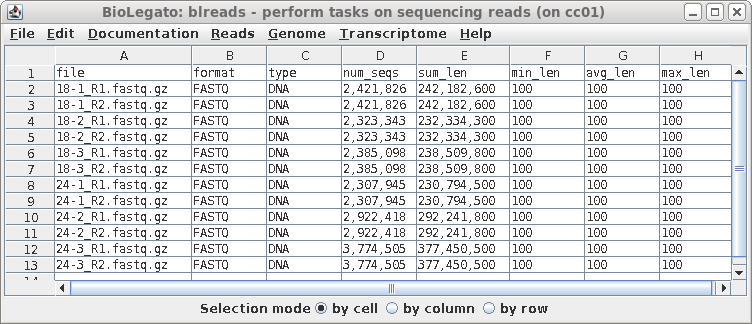
| In this case, all of the files look fairly
similar in terms of the number of reads and the average
length of reads. At this point, if there was a file that looked to be aberrant and you knew that it should be discarded, simply select the name of the bad file and delete the link using File --> Deletefiles. |
 |
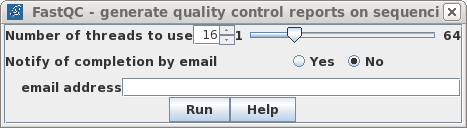
| Typical read files are larger than this
sample dataset, so with larger files, you may wish to speed
up FASTQC by setting a larger number of cores. In most
cases, results will appear soon enough that you don't need
to set notification of completion by email. Click on Run. |
 |
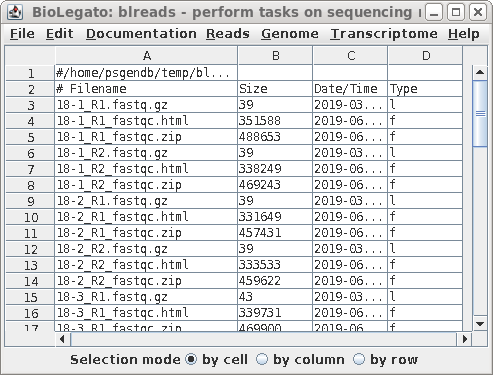
| blreads now lists the output files from
FASTQC. Files with the .html extension are viewable in any
web browser. The .zip files are the accompanying graphics
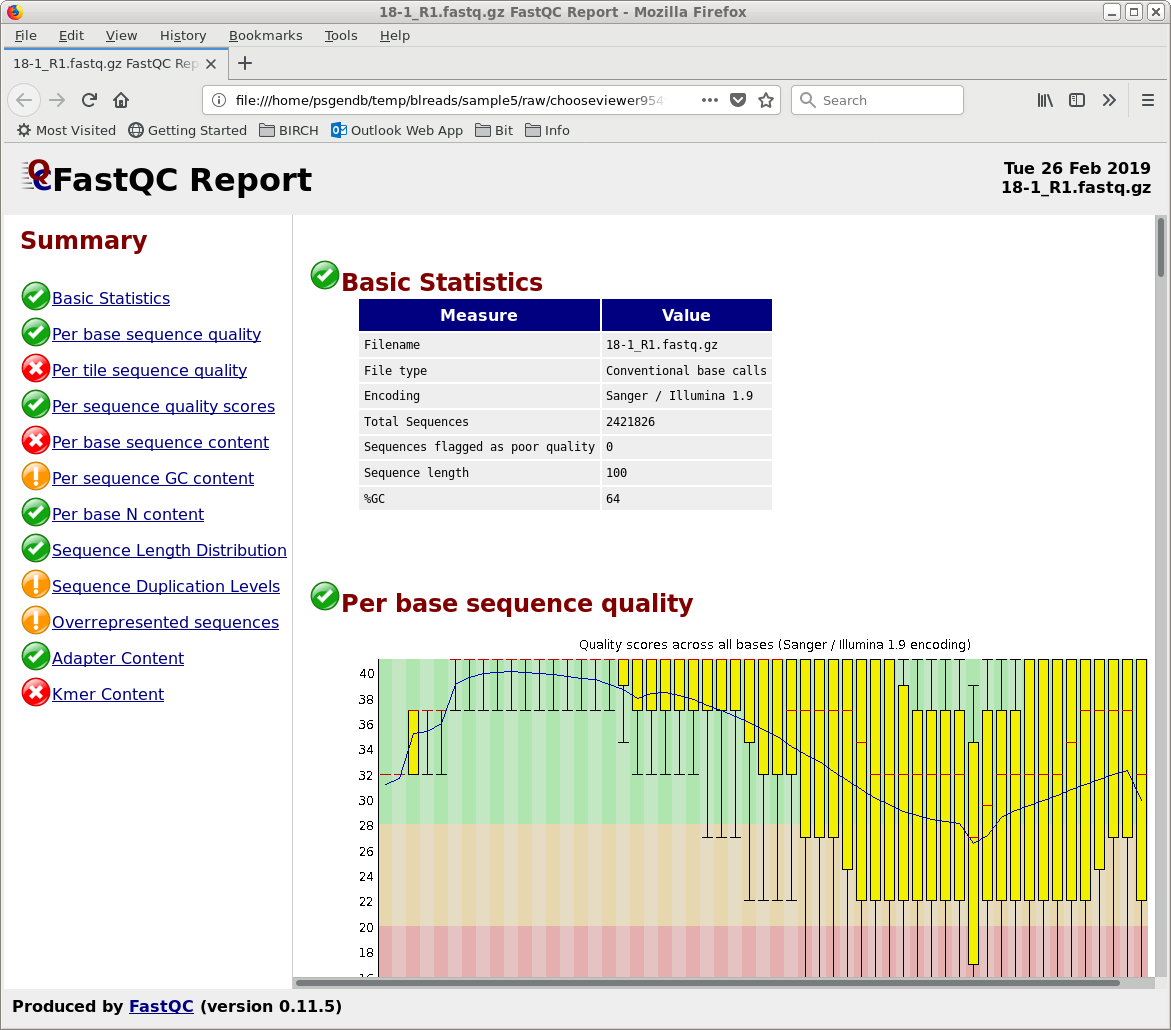
used by the web files. To view any report, select an HTML file and choose File --> View file. The HTML file will pop up in the browser. |
 |
| For large RNAseq datasets,
we have found that read files generated using trim_galore
cause Trinity, rnaspades and SOAPdenovo-Trans to terminate
without completing the assembly. Assemblies were successful
using reads corrected by Trimmomatic. Therefore, this
tutorial will use Trimmomatic. Trimmomatic is more complex
to use than trim_galore, because it has a wider array of
choices for read trimming.. Trimmomatic can be run from
blreads using Reads --> Trimmomatic. |
| Most Illumina services use R1 and R2 as the
substrings which distinguish the left and right read pair
files for a given library, so by default, R1 and R2 is set
for "unique string for left/right reads" For this dataset, all read files use the fastq.gz extension. (By default this will be set to .fq.gz). In some cases, you may need to specify more than one set of file extensions as a comma-separated list eg. .fq,.fastq Clicking on the Hints button will give a more detailed explanation of these parameters. |
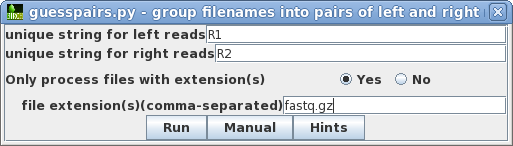 |
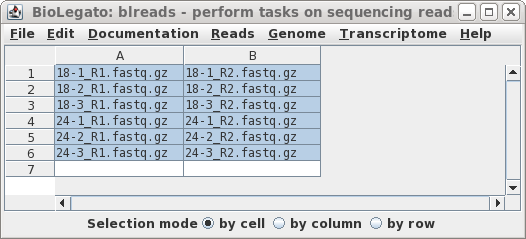
| Clicking on Run will bring up a new
blreads window with the best quess of file pairing, in two
columns. Usually, guesspairs.py gets it right. Files for
which a pair cannot be found (ie. single-end reads) would be
listed in a single column. To run Trimmomatic with these read pairs, choose Edit --> SelectAll, and then Reads --> Trimmomatic. |
 |
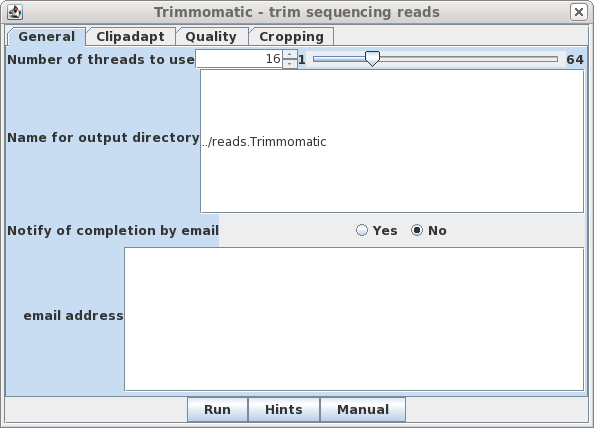
| General - The parameters for
Trimmomatic are grouped into four tabs. The General tab has
basic settings. By default, output will go to the
../reads.Trimmomatic directory. Trimmomatic performs each of the processing steps in an order specified by the user. Individual parameters can be turned on or off, and the order in which they are performed is set by the rank parameter. |
 |
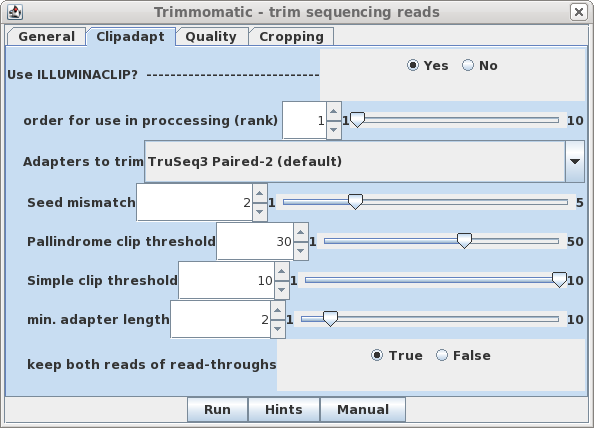
| Clipadapt - By default, the
ILLUMINACLIP step is performed first (ie. rank=1). The
defaults are those given in the Trimmomatic manual, with the
exception that keep both reads of read-throughs is
set to true. This may help to avoid cases where a single
read of a read pair is deleted during trimming, which can
cause some transcriptome assembly programs to crash. |
 |
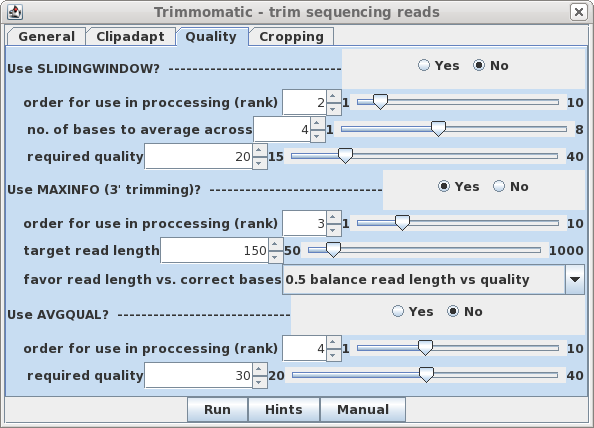
| Quality - By default, SLIDINGWINDOW
and AVGQUAL are off. Set MAXINFO to Yes. Since rank is set
to 3, we don't need to change this for MAXINFO to be
performed as the next step. |
 |
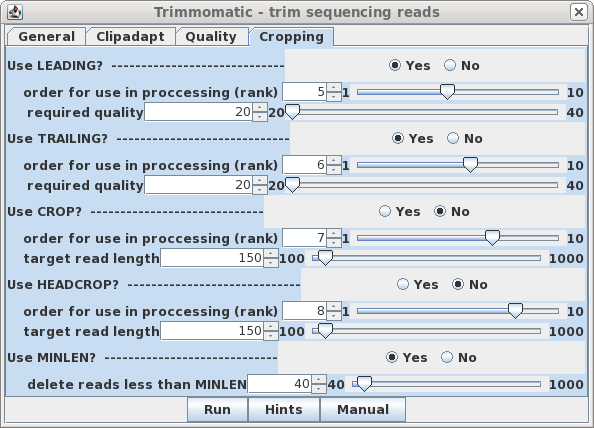
| Cropping - It's a good idea to eliminate poor
quality nucleotides from 3' and 5' ends of reads, so set
LEADING and TRAILING to Yes. Finally, the last step is MINLEN, which eliminates read pairs below a specific length. For 100 bp reads a value of 40 is a reasonable compromise between very short reads, which would be difficult or impossible to uniquely map to transcripts, and much longer reads, which might compromise coverage of reads located in the 5' ends of transcripts. No rank is assigned, because this step must be done after all other trimming steps are completed. |
 |
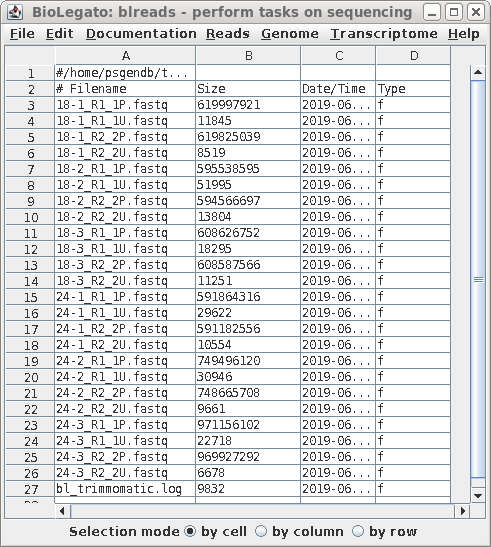
| When Trimmomatic has processed all files, a
new blreads window will pop up. It is important to
note that this new window is running in the
reads.Trimmomatic directory. At this point, one can probably
close the previous blreads window, which is running in the
raw directory. The next steps will be done in the
reads.Trimmomatic directory. (Optional: You may wish to run FASTQC at this point to verify that the properties of the processed reads are similar to the original reads.) For each raw read file, there are now two output files:
|
 |