TUTORIAL: Generating Gene Expression data from RNAseq reads |
June 29, 2020 |
TUTORIAL: Generating Gene Expression data from RNAseq reads |
June 29, 2020 |
| Pre-processing
and assembly of reads A simplified workflow for transcriptome assembly is shown at right. Transcriptome assembly is carried out within the transcriptome directory, and for each major step, a separate subdirectory is used. Raw read files are saved in the reads directory, and symbolic links to these files, with short, meaningful names, are created. Sequencing adaptors are removed from the raw reads by Trimmomatic and the files containing the trimmed reads are saved in reads.Trimmomatic. The trimmed reads are used as input by Rcorrector, which corrects errors in the reads and writes the corrected reads to the reads.Trimmomatic.Rcorrector directory. At each step, FASTQC is run to check the properties of the reads. Finally, the assembly itself is done using programs such as Trinity which produces contig files and scaffold files. For each assembly, transrate produces a reports with extensive statistics that can guide the choice of which assembly is best, or which assemblies should be repeated with different a parameters for improvement. |
| Generation of gene
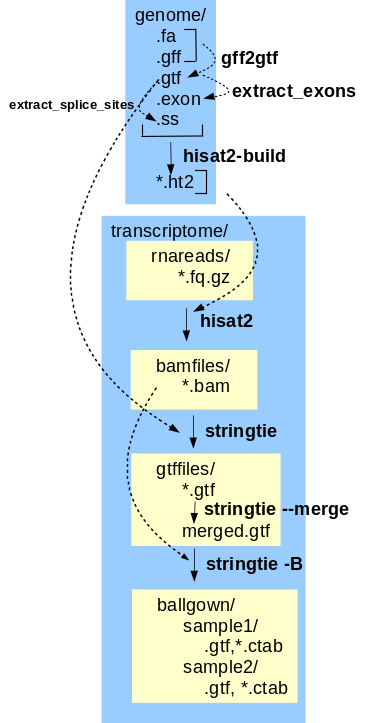
expression data The final transcriptome assembly can now be used to generate gene expression data from the corrected reads. This tutorial implements the Hisat, Stringtie, Ballgown gene expression pipeline. Hisat-build creates files that index the features in the annotated genome, so that reads can be mapped to features. Mapped reads are written to .bam files. For each read file, hisat2 maps reads to the genome. Next, stringtie assembles the reads into transcripts, including alternative splicing products. Transcripts mapped to genomic locations are written to .gtf files. Transcripts from all sets of reads are merged into a single .gtf file by stringtie --merge. The final step is to generate tables of gene expression values. stringtie -B reads the mapped reads from the .bam files and the mapped transcripts from the .gtf files, and writes tables of FPKM values to comma-separated value files that can be read by the BioConductor R package for further analysis. |
 |