TUTORIAL:
Generating
Differential Expression Data from Reads
|
June 19, 2019 |
TUTORIAL:
Generating
Differential Expression Data from Reads
|
June 19, 2019 |
| Rhodia1_1_AssemblyScaffolds.fasta |
361 genome scaffolds in FASTA format |
| Rhodia1_1_GeneCatalog_20180403.gff3 |
feature annotation in GFF3 format |
We will use the read files by creating symbolic links to the ones created in the previous tutorial on Read Correction. If you have not done that tutorial, you can also download gzipped files containing the corrected reads.
In your de directory,
mkdir reads
cd reads
To create symbolic links to existing read files:
cd reads
Start blreads in the de/reads dir
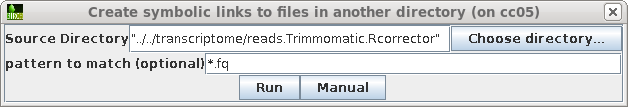
Use the directory chooser to select the name of the directory containing the corrected reads. The chooser will use the fully-qualified path to the files for the symbolic links. The link names can be made shorter by editing the Source Directory line to a relative path ie.
../../transcriptome/reads.Trimmomatic.Rcorrector
In either case, set the pattern to "*.fq" and click on Run.
The blreads window will refresh, showing the symbolic links to the read files.
Next, download the .fq.gz files found at ftp://ftp.cc.umanitoba.ca/psgendb/BIRCH/data/blreads/de/reads.Trimmomatic.Rcorrector . The files contain the corrected reads, which should be saved to your reads directory. The total size of these files is around 2.3 Gb.
Next unzip the files, using either unpigz (faster) or gunzip:
unpigz *.gz
or
gunzip *.gz
Verify that the files have been unzipped correctly:
ls -l
total 8623152
-rw-rw-r-- 1 psgendb psgendb 654227590 Mar 21 13:27 18-1_R1_val_1.cor.fq
-rw-rw-r-- 1 psgendb psgendb 657474671 Mar 21 13:27 18-1_R2_val_2.cor.fq
-rw-rw-r-- 1 psgendb psgendb 631270036 Mar 21 13:31 18-2_R1_val_1.cor.fq
-rw-rw-r-- 1 psgendb psgendb 630134395 Mar 21 13:31 18-2_R2_val_2.cor.fq
-rw-rw-r-- 1 psgendb psgendb 645589447 Mar 21 13:36 18-3_R1_val_1.cor.fq
-rw-rw-r-- 1 psgendb psgendb 647000420 Mar 21 13:36 18-3_R2_val_2.cor.fq
-rw-rw-r-- 1 psgendb psgendb 632249759 Mar 21 13:40 24-1_R1_val_1.cor.fq
-rw-rw-r-- 1 psgendb psgendb 631184299 Mar 21 13:40 24-1_R2_val_2.cor.fq
-rw-rw-r-- 1 psgendb psgendb 799667084 Mar 21 13:45 24-2_R1_val_1.cor.fq
-rw-rw-r-- 1 psgendb psgendb 798155913 Mar 21 13:45 24-2_R2_val_2.cor.fq
-rw-rw-r-- 1 psgendb psgendb 1035312919 Mar 21 13:51 24-3_R1_val_1.cor.fq
-rw-rw-r-- 1 psgendb psgendb 1033130655 Mar 21 13:51 24-3_R2_val_2.cor.fq
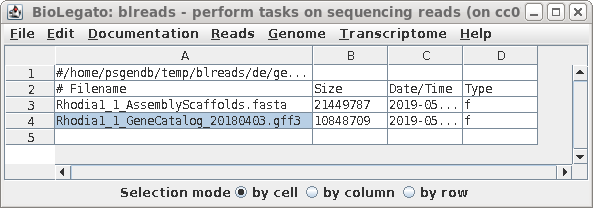
| Go to the de/genome directory and launch
blreads. Select the GFF3 file as shown. |
 |
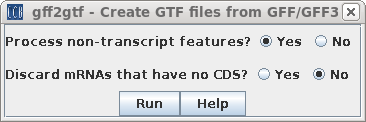
| Next, choose Transcriptome --> gff2gtf.
This command uses gffread to convert gff or gff3 files into
the GTF format needed by Hisat2. You may wish to set the two parameters which govern which annotations go into the GTF file. In this case choose "Yes" for Process non-transcript features. |
 |
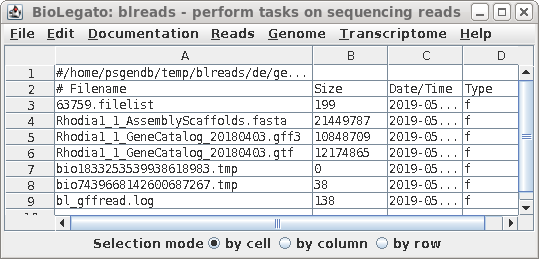
| The blreads window will be refreshed to show
that the GTF file has been created. |
 |
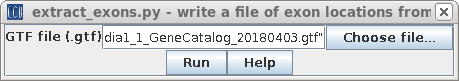
| Hisat2 requires separate files for annotation
of exons and splice sites. Choose Transcriptome -->
extract_exons.py and choose the GTF file. |
 |
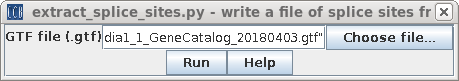
| Next, choose Transcriptome -->
extract_splice_sites.py and again choose the GTF file. |
 |
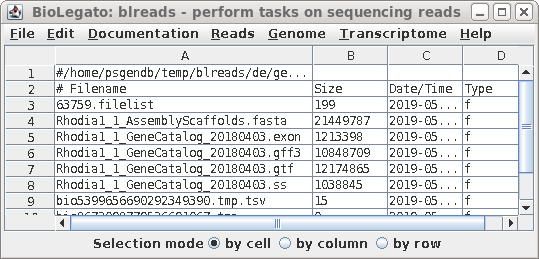
| Now, the exon (.exon) and splice site (.ss)
files should also be visible in blreads. |
 |
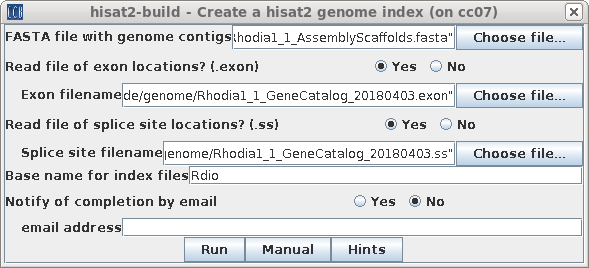
| Choose Transcriptome --> hisat2-build.
Select Rhodia1_1_AssemblyScaffolds.fasta, and the exon and
splice site files. Remember to click "Yes" to tell blreads
to use the exon and splice site files. Click on Run to start hisat2-build. |
 |
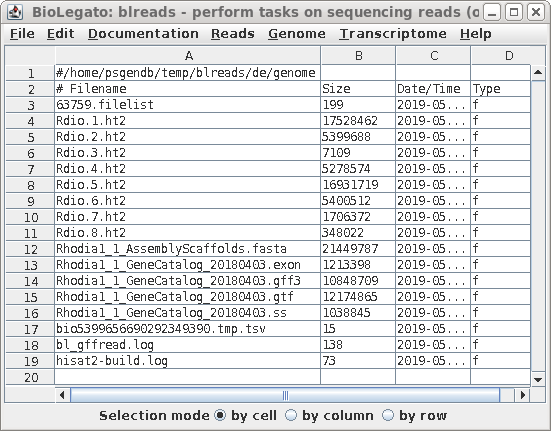
| A new blreads window will appear showing the
creation of eight .ht2 files, which will be used in
subsequent steps. |
 |
| The next step is to create .bam files which contain reads mapped to the genome. Because this step utilizes the corrected reads, we need to run blreads in the de/reads.Trimmomatic.Rcorrector directory. Use guesspairs.py to group read pair files, as shown: | 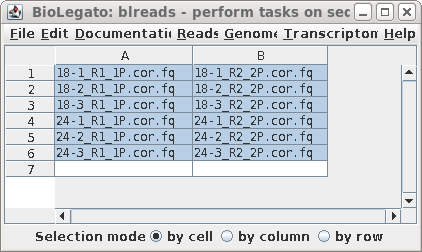 |
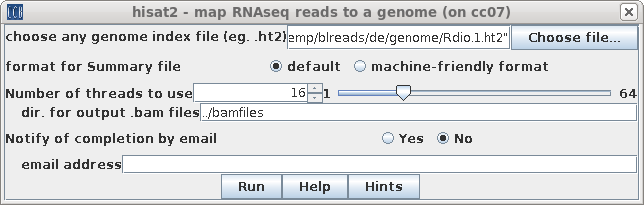
| Choose any of the .ht2 files (eg.
Rdio_1.ht2). Output will be written to de/bamfiles, by default. Click on Run. |
 |
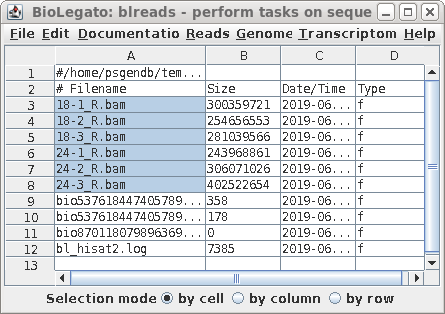
| For each pair of read files, hisat2 will
write a .sam file mapping the read pairs to the genome.
Because stringtie will require the reads to be sorted by
position in the reference genome, the .sam files are read
and sorted by samtools, and output written to .bam files,
which contain the same information in smaller binary format.
The .sam files are deleted. A new blreads window will open
in the bamfiles directory. |
 |
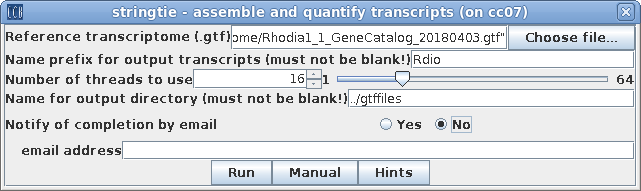
| Stringtie assembles the reads on to
transcripts, mapped to the annotated genome. First, make sure that the .bam files from the previous step are all selected. Next, choose Transcriptome --> stringtie. Choose the RTF file created in step 1 as the reference transcriptome. Type in a prefix which will be part of the name of each transcript written to the output. These names will be used in all downstream analysis, so make it meaningful, but short. By default, output will be written to a directory called de/gtffiles. |
 |
| When stringtie is finished, for each .bam file in the bamfiles directory, there is now a GTF file in the gtffiles directory. | 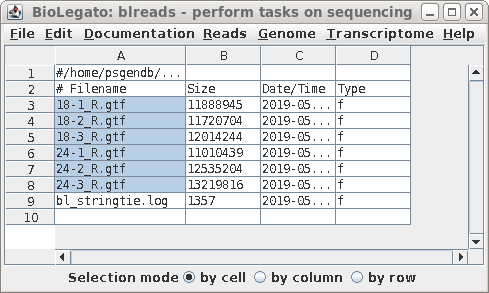 |
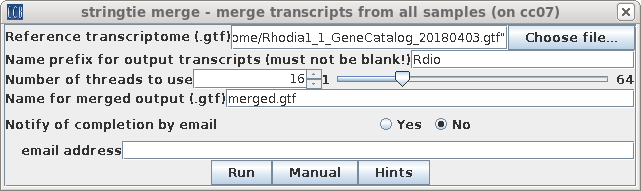
| Next, we need to create a merged file
containing the information from all of the GTF files. Select
all of the GTF files produced in the previous step and
choose Transcriptome --> stringtie merge. Again, we chose the genome GTF file as the reference transcriptome, and set the prefix to "Rdio". Output will be written to the gtffiles directory to a file called merged.gtf. |
 |
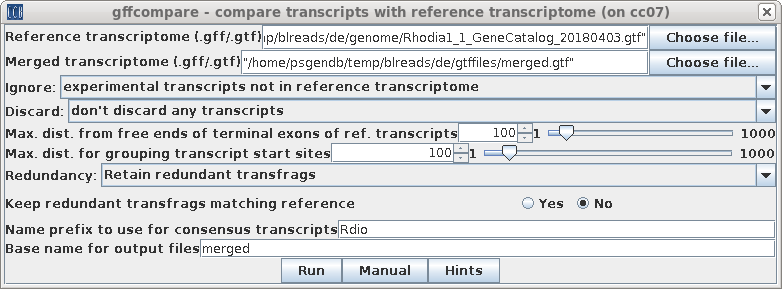
| To run gffcompare, choose Transcriptome
--> gffcompare. Again, select the GTF file for the reference genome, and the merged transcriptome file, merged.gtf from the gtffiles directory. Select Ignore: experimental transcripts not in the reference genome. Once again, specify "Rdio" as the prefix for transcripts. Click on Run. |
 |
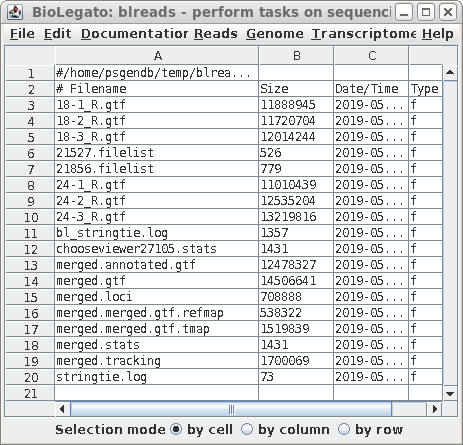
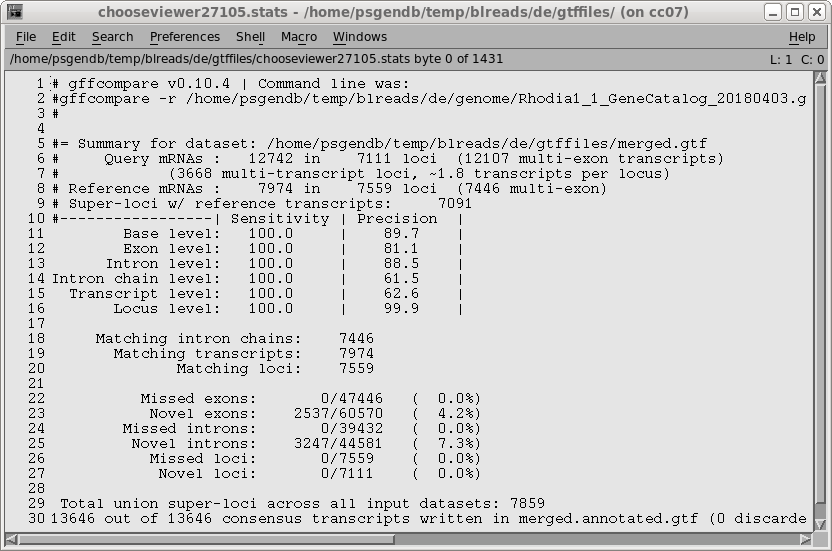
| A new blreads window will pop up in the
gtffiles directory. Merged.annotated.gtf contains the transcripts from merged.gtf, minus the novel experimental transcripts. |
 |
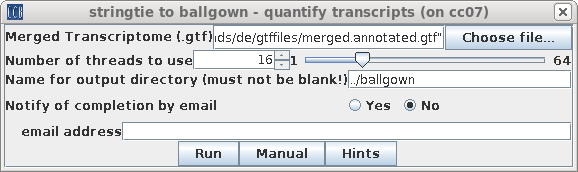
| Because we now want to use the reads to
measure gene expression levels for the annotated
transcripts. Open blreads in the bamfiles directory, select all .bam files and choose Transcriptome --> stringtie to ballgown, and set the merged transcriptome file to merged.annotated.gtf. |
 |
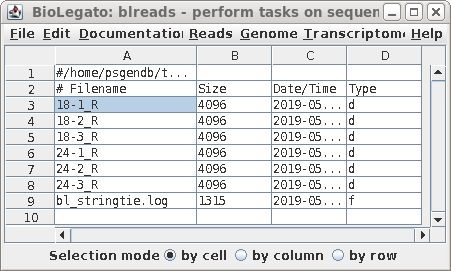
| The results will be written to the ballgown
directory. Note that almost all of the contents of the ballgown directory consists of subdirectories, one for each .bam file. The contents of each subdirectory can be viewed in either a file manager, or in blreads. For example, to view the contents of 18-1_R, choose 18-1_R, and then File --> Open directory. |
 |
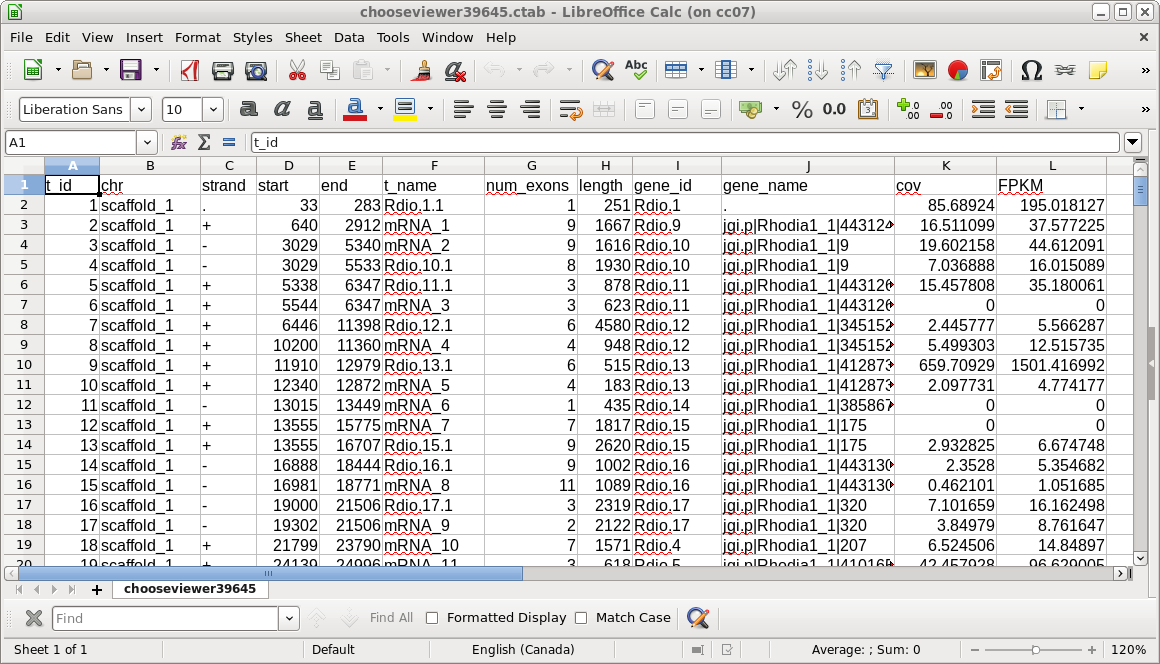
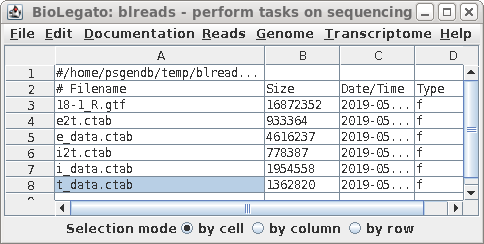
| Each directory has a gtf file for one set of
reads, as well as a number of CSV files with the .ctab file
extension. The most important one is t_data.ctab, which has the FPKM calculations for each transcript. You can view this file by selecting t_data.ctab, and then File --> View file. |
 |