BioLegato:
|
June 2, 2018 |
| Object-oriented
graphic interfaces - BioLegato borrows the concept of
objects from object-oriented databases and
object-oriented programming languages. |
birch
- launcher for BIRCH GUI programs bldna - DNA/RNA sequences blprotein - amino acid sequences blnalign - work with DNA/RNA sequence alignments blpalign - work with protein sequence alignments bltable - spreadsheet data blreads - perform tasks on sequencing reads blmarker - molecular marker data blncbi - search NCBI databases blnfetch - retrieve DNA or RNA sequences from NCBI blpfetch - retrieve amino acid sequences from NCBI bltree - display and manipulate phylogenetic trees birchadmin - manage a BIRCH site blgeneric - empty BioLegato instance |
birch -
BIRCH launcher
birch is a BioLegato interface for launching GUI programs in the BIRCH system. In addition to launching the other BioLegato interfaces, birch organizes the main BIRCH programs into categories, making it easy to discover which programs are available on the system.
birch is a BioLegato interface for launching GUI programs in the BIRCH system. In addition to launching the other BioLegato interfaces, birch organizes the main BIRCH programs into categories, making it easy to discover which programs are available on the system.
bldna - DNA sequence data
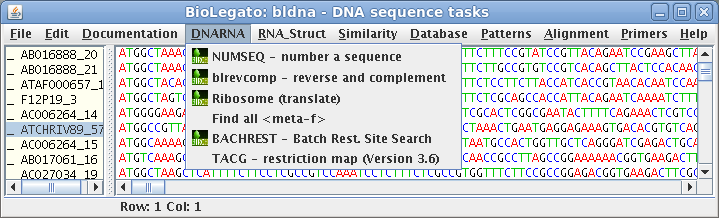
| bldna is a BioLetgato instance
specialized for working with dna sequence data. The
sequence canvas is designed as a multiple sequence
alignment editor allowing the insertion and deletion of
gaps. As an alternative to using the entire alignment,
specific sections of the alignment can be selected in
blocks and used for further analysis. |
 |
To launch bldna from the command line, type
bldna
The File --> Open menu can read four types of files: GDE format, GDE flatfile, FASTA and GenBank. Many other sequence formats can be imported using File --> Import Foreign Format.
The GDE flat file format was designed to allow "sequences" to contain plain text. This was originally intended as a commenting feature, but we can take advantage of it for reading many kinds of text data. Four additional BioLetgato interfaces exploiting this feature are described below.
blprotein - protein sequence data
| blprotein is a BioLetgato instance specialized for working with protein sequence data. It has the the same kind of sequence canvas as bldna. blprotein also uses the same methods as bldna for file input. | 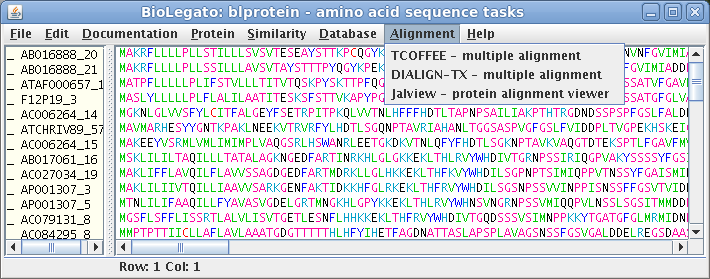 |
To launch blprotein from the command line, type
blprotein
blnalign - work with aligned DNA or RNA sequences
| blnalign is a BioLetgato instance that performs tasks on a set of aligned DNA or RNA sequences. Tasks include various ways of displaying alignments, and construction of phylogenetic trees. | 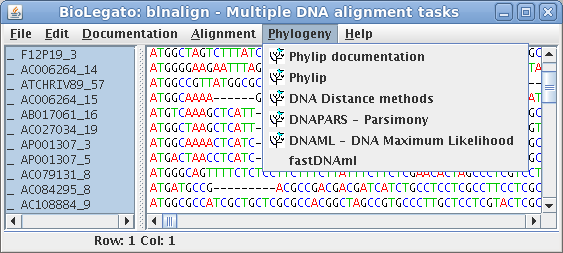 |
To launch blnalign from the command line, type
blnalign
blpalign - work with aligned protein sequences
| blpalign is a BioLetgato instance that performs tasks on a set of aligned amino acid sequences. Tasks include various ways of displaying alignments, and construction of phylogenetic trees. | 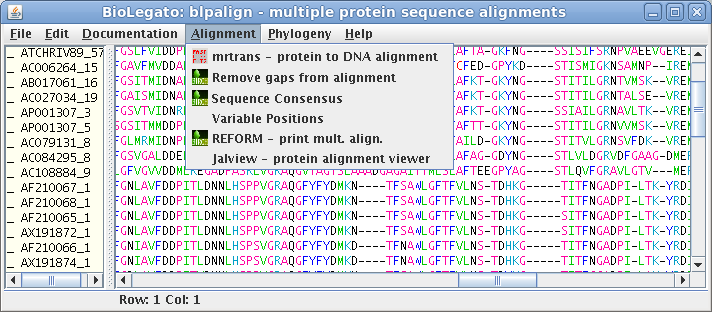 |
To launch blpalign from the command line, type
blpalign
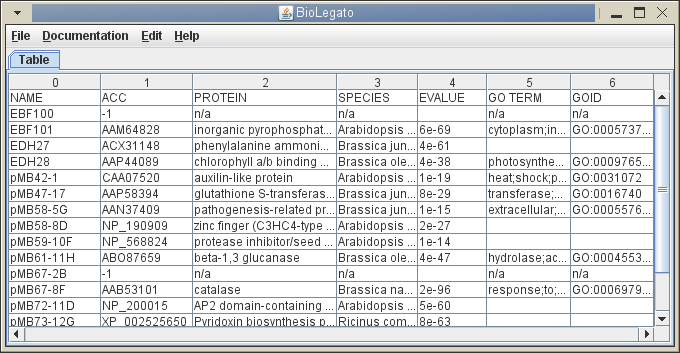
bltable - Spreadsheet Data (eg. Accession numbers, TaxID numbers, GI numbers)
The bltable interface is has a minimal set of functions, and is not meant to be a fully-featured spreadsheet. Rather, it can be thought of as generic table application that can be the starting point for creating spreadsheet applications for specific types of data. In OO terms, bltable is like an abstract class that can be extended to produce a more elaborate class.
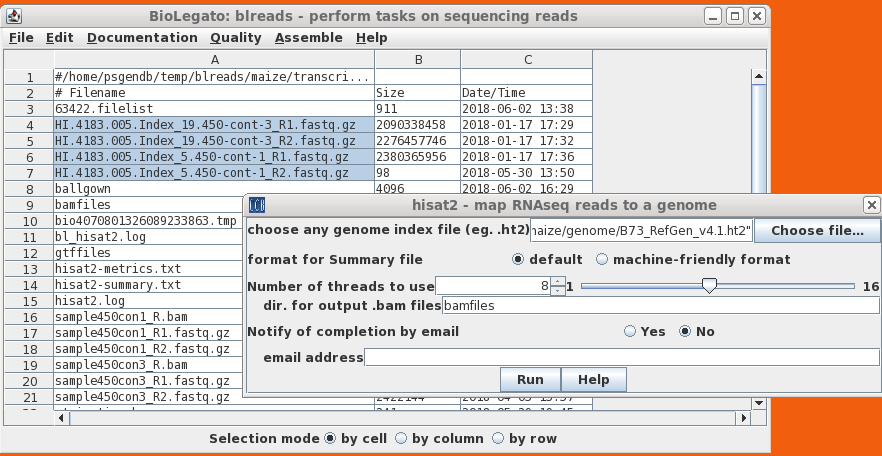
blreads - perform tasks on sequencing reads
blreads manages the many tasks on NGS
sequencing data, including:
|
 |
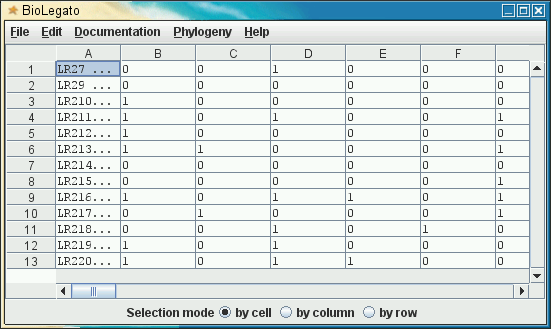
blmarker - Marker Data
blmarker is a BioLetgato interface specialized for molecular marker data, such as microsatellites, AFLPs, SRAPS, or many other types of molecular markers. |
 |
To launch blmarker, type
blmarker
At present, blmarker can read marker data in Phylip Discrete Data format or from comma-separated value (.csv) files, as generated from a spreadsheet. This format takes 1's and 0's as markers, usually representing presence or absence of a marker band. To read in discrete data, choose File --> Import Phylip Discrete Data.
blncbi - Search NCBI databases
blncbi is a BioLegato interface for searching NCBI databases and retrieving results.
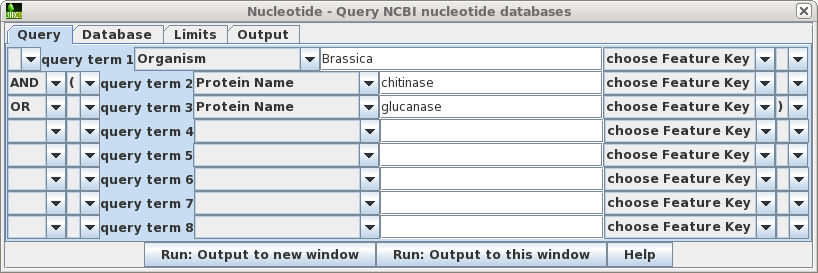
Above is shown the query builder for querying the nucleotide databases. In this example, we are searching for any sequences from the genus Brassica, for which the Protein Name field is set to either chitinase or glucanase. Note that parentheses and logical operators can be specified using pull-down menus. This search is equivalent to the NCBI Entrez term:
Brassica [ORGN] AND (chitinase [Protein Name] OR glucanase [Protein Name])
The results are returned to a new blncbi window:
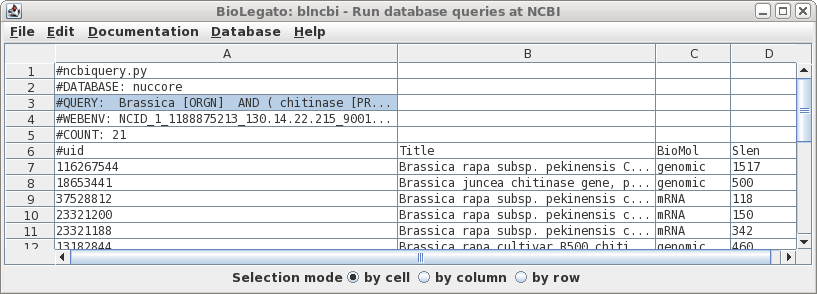
The results in this table can be used for subsequent searches that narrow the set of results, or to retrieve sequences from the NCBI database.
To launch blncbi, type
blncbi
blncbi can read and write tab-separated value (.tsv) files.
blnfetch - Retrieve DNA or RNA sequences from a remote database
blnfetch is a BioLetgato interface specialized for retrieval of DNA or RNA sequences, using NCBI GI numbers or ACCESSION numbers as input. At right, we see a blnfetch window generated from a blastn search by bldna. The NCBI database includes an increasing number of chromosome-sized entries. SeqFetch allows the user to select the minimum and maximum length of sequences retrieved, making it easy to eliminate very large sequences. |
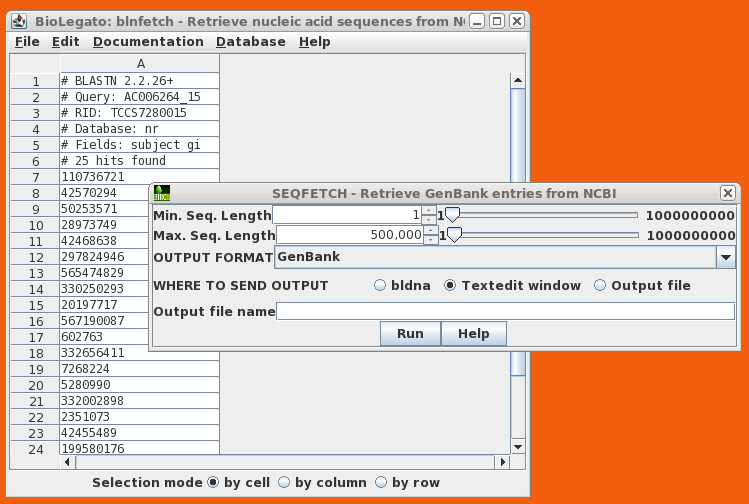 |
To launch blnfetch, type
blnfetch
At present, blnfetch can read lists of NCBI GI or ACCESSION numbers from a comma-separated value (.csv) file, for example, as generated from blast search results. To read in ID data, choose File --> Import Data from CSV file.
blpfetch - Retrieve amino acid sequences from a remote database
blpfetch is a BioLetgato interface specialized for retrieval of amino acid sequences, using NCBI GI numbers or ACCESSION numbers as input. At right, we see a blpfetch window generated from a blastp search by blprotein. |
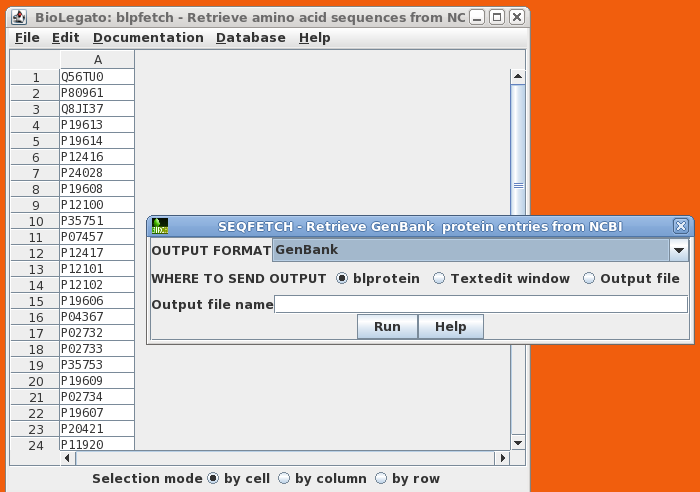 |
To launch blpfetch, type
blpfetch
At present, blpfetch can read lists of NCBI GI or ACCESSION numbers from a comma-separated value (.csv) file, for example, as generated from blast search results. To read in ID data, choose File --> Import Data from CSV file.
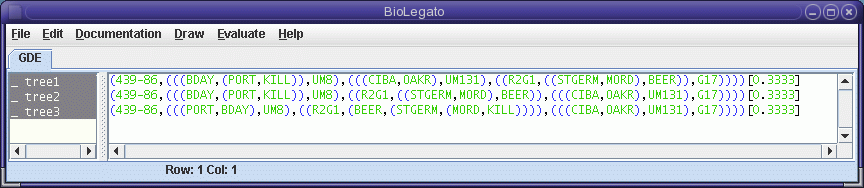
bltree - Phylogenetic Tree Data
bltree is a BioLetgato interface specialized for phylogenetic tree data. To launch bltree type
bltree
At present, bltree can only read trees in Phylip tree format. To read in a file containing trees, choose File --> Import Trees.
birchadmin - BIRCH Administration Tool
birchadmin is a BioLetgato interface for managing a BIRCH site. To launch birchadmin type
birchadmin
or in birch, choose File --> birchadmin.

(Note: you must be logged in as the BIRCH administrator to run birchadmin.)
blgeneric - empty Biolegato instance
blgeneric is a BioLetgato interface that launches BioLegato without any menus or canvas. This is mainly for demonstration purposes, to illustrate the fact that almost all functionality of BioLegato is programmable. In the terminology of Object-Oriented programming, think of BioLegato as an abstract class that is extended to create real classes. So in a way blgeneric is like instantiating an abstract class. To launch type 'blgeneric'.
|
|
    |
|