Lecture 6, part 1 of 2
| PLNT4610/PLNT7690
Bioinformatics Lecture 6, part 1 of 2 |
REFERENCES
Baxevanis AD (1998) Practical Aspects of Multiple Sequence Alignment. In Baxevanis AD and Ouellette BFF. Eds. Bioinformatics: A practical guide to the analysis of genes and proteins. John Wiley, Toronto.
Setubal, J. and Meidanis, J. (1997) Introduction to Computational Molecular Biology. PWS Publishing Co., Toronto. Ch. 3 "Sequence Comparison and Database Search".
Lectures by Dr. Michael
Zuker, Bio-5495, Institute for Biomedical Computing, Washington
University Medical School
[http://www.genetics.wustl.edu/bio5495/1999-course/lecture.7/
]
G. Fuellen, Multiple Alignment. Complexity International 4, 1997. URL http://journal-ci.csse.monash.edu.au/ci/vol04/mulali/mulali.html.
Toompa, M.
(2000) Multiple sequence alignment
http://www.cs.washington.edu/education/courses/527/00wi/lectures/lect06.pdf
10 20 30 40 50 60
consensus -----xxxxx-xxxxlxxxxlxxxxxxxsxaxgIxiYWGQngnEGsLadtCxtgnYxxVn
CACHIT .....me--k.cfniipsll.islliks.n.a..av...........q.a.n.n..qf..
PSTCHIT .....meslk.kaslvlfpi.vlslfnh.n.a..av......g........n....ef..
NTACIDCL3 ....mi----.kysf.ltalvlflralkle.gd.v................a.n..ai..
S66038 .maaki----.vsvlflisl.ifasfes.hgsq.v.......d........ns...gt.i
CUSSEQ_1 .....maahkiittt.siff.lssifrs.n.a..a.............s..a....ef..
CUSSEQ_2 .....maahk.ittt.siff.lssifrs.d.a..a.............s..a....ef..
CUSSEQ_3 .....maahk.ittt.siff.lssifhs.d.a..g.............s..a....ef..
VIRECT .....-maclkqvsa.llpl.fisffkp.h.g..sv.............a.n....ky..
VURNACH3A .....-----.----------------------.sv.............a.n....ky..
ATHCHIA ..mtnmtlrkhviyf.ffiscslskpsdasrg..a..........n.sa..a..r.ay..
VURNACH3B mvktkisl--.llpl.ff---tlvgtsha--g..a..........t.sea.d..r.th..
NTBASICL3 .mnikvsl--lfilpifl----llltskvk.gd.vv....dvg..k.i...ns.l.ni..
70 80 90 100 110 120
consensus iAFlsxFGxgQtPxlNLAGHCnPxxnxCxxxsxxixxCqsxgiKvllSxGGgxgxYslxS
CACHIT .....t..n..n.qi......d.st.g.tkf.pe.qa..ak.......l...a.s...n.
PSTCHIT .....t..s....q.......d.ss.g.tgf.se.qt..nr.......l..sa.t...n.
NTACIDCL3 ....vv..n..n.v.......d.naga.tgl.nd.ra..nq....m..l...a.s.f.s.
S66038 l..vat..n....a.......d.-atn.nsl.sd.kt..qa.......i...a.g...s.
CUSSEQ_1 .....s..s....v.........dn.g.afv.de.ns...qnv.....i...v.r...s.
CUSSEQ_2 .....s..s..a.v.........dn.g.afl.de.ns.k.qnv.....i...a.s...s.
CUSSEQ_3 .....s..g....v.........dn.g.til.ne.ns...qnv.....i...t.s...y.
VIRECT ....ft..g....q.........si.n.nvf.dq.ke...kd......l..as.s...t.
VURNACH3A .....a..g....q.........si.n.nvf.dq.kg...r.....p.l..as.s...s.
ATHCHIA v...vk..n....e.........aa.t.thfgsqvkd...r....m..l...i.n..ig.
VURNACH3B ....nk..n....em........at.s.tkf.aq.ky...kn......i...i.t.t.a.
NTBASICL3 .....s..nf...k.......e.ssgg.qqltks.rh...i...im..i...tpt.t.s.
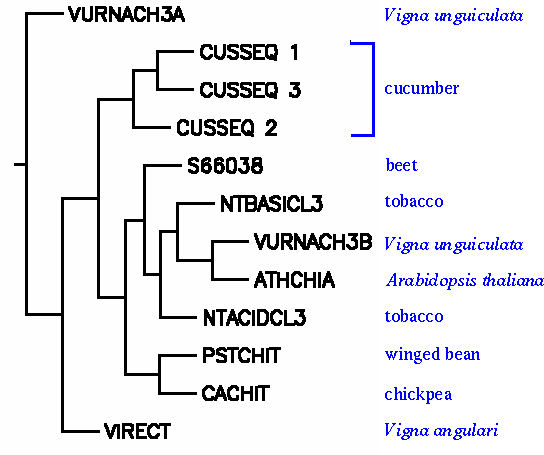
However, relationships among the chitinase III proteins are easier to visualize in a phylogenetic tree. For example, we see that among the legumes, chitinase III from winged bean and chickpea are closely-related, but sequences from other legumes in the genus Vigna appear to have evolved from some more distant ancestor. For example, VURNACH3B clusters with the crucifer Arabidopsis.
Perhaps the biggest problem concerning multiple alignment and phylogeny are that they are interdependent. In principle, multiple alignment could be done in the absence of any knowledge of the evolutionary relationships of the species or proteins being aligned. In practice, exhaustive methods for mulitple alignment scale on a very sharp exponential curve, so that exhaustive alignments with more than a few sequences are impossible. Alignments can be built quickly using phylogenetic trees and pairwise comparisons as guides. The price we pay is the loss of independence. The alignment depends on some sort of phylogeny, while the phylogeny must be calculated from an alignment.
A good compromise is to do the protein alignment, and from the protein alignment, construct a DNA alignment for use in the phylogeny. The pal2nal.pl script can do this.
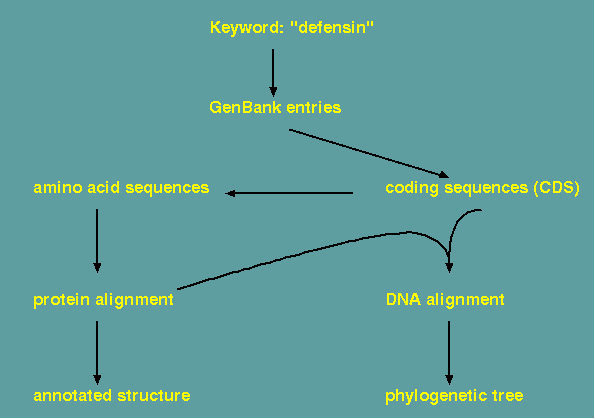
 Implementation of such a workflow
might be done in a number of ways. There is no one protocol for
constructing alignments and phylogenies. At each step, decisions
must be made as to which approach to take. In some cases, it may
be necessary to try several methods before choosing one. As
well, results at one step often make it necessary to go back
several steps and refine the dataset. For example, a poor
phylogeny may indicate the need to re-do the alignment, and then
to retry the phylogeny.
Implementation of such a workflow
might be done in a number of ways. There is no one protocol for
constructing alignments and phylogenies. At each step, decisions
must be made as to which approach to take. In some cases, it may
be necessary to try several methods before choosing one. As
well, results at one step often make it necessary to go back
several steps and refine the dataset. For example, a poor
phylogeny may indicate the need to re-do the alignment, and then
to retry the phylogeny.
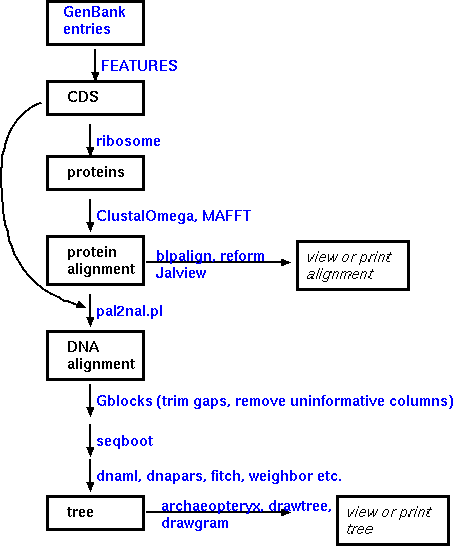
The steps in constructing an alignment and a phylogeny are illustrated using programs from the BIRCH system. Assume a set of GenBank entries has been retrieved, all of which represent homologous genes from several species. In GenBank entries, protein coding sequences are annotated as 'CDS' features. The FEATURES program can extract CDS sequences from a group of GenBank entries automatically. Next, the coding sequences must be translated. Two multiple alignment program are available. ClustalOmega and MAAFT. To display the alignment for evaluation or final publication, it is best to try several of the programs listed to tailor the output to your needs. REFORM generates straight ASCII text, which can be easily imported into a word processor or drawing program. The other programs listed generate PostScript output for direct viewing or printing.
If an aligned DNA sequence is desired, pal2nal.pl can read in an aligned protein sequence and the corresponding DNA sequence and generate a file containing the DNA alignment.
The most complex part of the decision process involves choosing a strategy for phylogeny constructon. The three main phylogeny methods, parsimony, distance, and maximum likelihood, will be explained elsewhere.
Though not shown in the
figure, protein sequences can be directly used in phylogeny
construction. In this example, though, a phylogenetic tree is
constructed from the aligned DNA. The quality of the alignment
may be evaluated by bootstraping the alignment and rerunning the
phylogeny search. although this option is not usually possible
for large numbers of sequences when using a
computationally-intensive maximum likelihood program.
| PLNT4610/PLNT7690
Bioinformatics Lecture 6, part 1 of 2 |